Thinking outside the box to fight back neurodegeneration
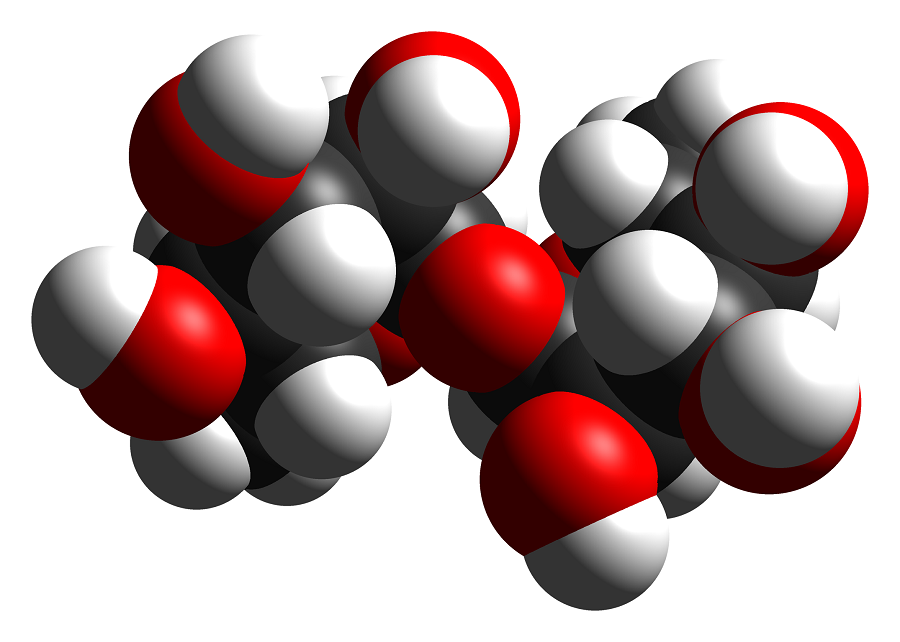
Neurodegeneration in a disease called mucopolysaccharidoses IIIB (MPS IIIB) results from the abnormal accumulation of essential molecules called mucopolysaccharides. In his lab at Baylor College of Medicine, Dr. Marco Sardiello and his colleagues have been looking to find alternative therapeutic strategies for this rare genetic disease. They recently discovered that a simple sugar, trehalose, might just be a part of it.

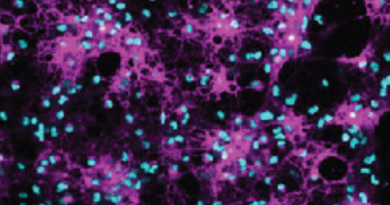
“MPS IIIB is one of about 50 lysosomal storage disorders characterized by the accumulation of material inside tiny cellular sacs called lysosomes,” said Sardiello, assistant professor of molecular and human genetics and a member of the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital and Baylor College of Medicine. “In the case of MPS IIIB, a mutation on a gene that codes for a lysosomal enzyme that breaks down a cellular material called heparan sulfate, renders the enzyme ineffective. Consequently, the lysosome cannot do its work of degrading heparan sulfate to either discard it or recycle it, and the material accumulates.”
Over the years, accumulation of heparan sulfate in lysosomes leads to degeneration of brain tissue. Although infants appear healthy at first, they slowly begin to show behavioral problems, hyperactivity, aggressiveness, sleep disturbances and loss of vision and hearing. Later in life, they become immobile and develop swallowing difficulties. Usually, they don’t live past the second decade.
Current strategies being tested for the treatment of this condition in animal models include attempting to correct the enzyme deficiency by providing a fully working enzyme. However, this approach faces challenges such as having limited ability to cross the blood-brain barrier and reaching brain areas where the enzyme is needed.
A non-traditional approach
“We explored a way to treat this condition with a nontraditional approach,” Sardiello said. “We recently discovered that the small sugar trehalose promotes the recycling of cellular waste. This made us think of an indirect approach to try to solve the accumulation of heparan sulfate.”
Instead of correcting the enzyme deficiency, we would try to overcome it by enhancing the cells’ natural ability to discard cellular waste,” Sardiello said.
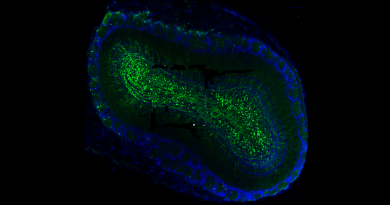
The researchers tested their approach on a mouse model of MPS IIIB. These mice have a mutation that results in the recapitulation of most of the clinical symptoms observed in patients, including progressive neurodegeneration, loss of vision, brain inflammation and shorter lifespan.
One group of MPS IIIB mice received trehalose in the drinking water, while another group of MPS IIIB mice did not receive the sugar. As controls, normal mice without the mutation were provided water with or without trehalose. The researchers observed the animals for 11 months, taking samples at the middle and at the end of the observation period.

“The results were highly encouraging,” said first author Dr. Parisa Lotfi, postdoctoral associate in the Sardiello lab. “The MPS IIIB mice treated with trehalose lived longer, improved their hyperactive behavior and delayed several neuropathological features, most notably the retinal degeneration, when compared with MPS IIIB mice not treated with trehalose.”
In addition, the researchers explored the molecular mechanism underlying the trehalose effect.
“Previously, we had discovered that trehalose activates TFEB, a master regulator of the lysosomal system. As the activity of TFEB increases, the degradation and clearance of molecules in the lysosomes becomes more efficient,” said Lotfi. “Here, we were the first to quantify the amount of trehalose that crosses the blood-brain barrier and activates TFEB in the brain. In turn, TFEB activated the lysosomal system, which led to enhanced clearance of material accumulation, reduced neuroinflammation, retinal degeneration and vision loss and extended lifespan.”
This study is the first to show that trehalose is acting through TFEB but no other molecules. Also, this is the first preclinical study showing that it is possible to delay retinal degeneration and loss of vision in a mouse model of MPS IIIB.
“The effect on the retina is important,” Lotfi said. “Loss of vision is one of the most devastating aspects of some lysosomal diseases. In animal models, treatments based on enzyme replacement therapy do not reach the brain and do not counteract loss of vision. We think that in the future our strategy can potentially be used either as the main therapy or complementary to other therapies.”
“Our results encourage us to consider that trehalose also may be effective in other lysosomal storage diseases, such as Batten disease,” Sardiello said. “Trehalose is a widely used food additive with no current restrictions for human use.”
Learn more about this work in the journal Autophagy
Other contributors to this work include Dennis Y. Tse, Alberto di Ronza, Michelle L. Seymour, Giuseppe Martano, Jonathan D. Cooper, Fred A. Pereira, Maria Passafaro and Samuel M. Wu. The authors are affiliated with one of more of the following institutions: Baylor College of Medicine, Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, the Institute of Neuroscience (Italy), Kings College London and the Hong Kong Polytechnic University.
This work was supported by the National Institutes of Health (NIH) grant NS079618, grants from the Team Sanfilippo Foundation and Swiss Sanfilippo Foundation and by the Brain Disorders and Development Training grant T32NS043124-13. This project was supported in part by the Hamill Foundation, by IDDRC grant number 1U54 HD083092 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development, a Shared Instrumentation grant from the NIH (1S10 OD016167) and by Hong Kong Polytechnic University grants G-YBQT and 1-ZE6H.d.