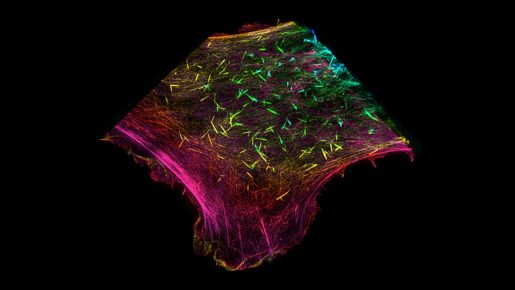
Changing the perspective on the ‘Cinderella of the cytoskeleton’
SETD2 is a protein well known as a chromatin remodeler, one that helps turn genes on or off by modifying histone proteins in the nucleus of the cell. When researchers discovered that SETD2 is mutated or lost in several cancer types, most commonly a type of kidney cancer called clear-cell renal cell carcinoma, all eyes turned toward SETD2 function in the nucleus of the cell to explain these cancers.
In 2016, the lab of Dr. Cheryl Walker, director of the Center for Precision Environmental Health at Baylor College of Medicine, made the unexpected discovery that SETD2 not only remodels chromosomes in the nucleus, but also microtubules of the cytoskeleton outside the nucleus. The cytoskeleton is a dynamic network of interlinking protein thread-like structures, including filaments and microtubules that extend throughout the cell. It gives a cell its shape and internal organization and provides mechanical support that enables cells to carry out essential functions like division and movement.

The Walker team found that SETD2 tags cytoskeleton microtubules with a methyl group. Loss of SETD2 resulted in defective delivery of chromosomes and problems with the separation of daughter cells during cell division.
“Our findings suggested that defects in SETD2 could not only affect gene expression but also functions controlled by the cytoskeleton, such as movement, metastasis and migration, which are very important for cancer cells,” Walker said. “We wondered whether SETD2 might target other cytoskeletal proteins.”
SETD2 works with Huntingtin and actin to regulate cell migration
Actin proteins, which form the filaments of the cytoskeleton, stood out as a prime target for SETD2. Two recent papers from the Walker lab have now revealed the role of SETD2 in modifying the actin cytoskeleton and its implications for two important functions of cancer cells, cell migration and autophagy.
One of the first findings was that SETD2 interacts with actin cytoskeleton and is able to modify actin in cells or in reactions using purified proteins. SETD2 adds three methyl groups to actin at a location called lysine-68. Interestingly, they found that SETD2 interacted with two other proteins to methylate actin in cells: Huntingtin (HTT) and the actin-binding adapter HIP1R.
Trimethylated lysine-68 regulates the normal dynamics of actin, including polymerization and depolymerization. Disrupting the SETD2-HTT-HIP1R association inhibited actin methylation, caused defects in actin dynamics and impaired cell migration, an important function of cancer cells.

These findings were very exciting because, to our knowledge, nobody had investigated the significance of the SETD2-Huntingtin interaction that had been known for over two decades,” said first and co-corresponding author Riyad Navroz Seervai, an M.D./Ph.D. student in the Medical Scientist Training Program who completed his Ph.D. thesis in the Walker lab.
“There was a limited list of papers about Huntingtin involvement in actin dynamics and cell migration, but enough to pursue the SETD2-Huntingtin-actin connection.”
Together, these data provided a new understanding of how defects in SETD2 and HTT can drive disease via disrupting cytoskeletal methylation and defects in cell migration. The researchers also were able to manipulate the SETD2-HTT-actin axis to show that changes in cell migration are specific to this new target of SETD2 (actin) rather than to chromatin or microtubules.
Read all about this work in the journal Science Advances.
A role for SETD2 in autophagy
The group also explored the influence of SETD2 on autophagy, a mechanism cells use to remove unnecessary or dysfunctional components.
“Dr. Walker’s lab has an extensive background and expertise in studying autophagy,” Seervai said. “There was always a suspicion that SETD2 might be involved in this process, but it had not been tested. This project got off the ground once we performed the initial experiments looking at autophagy markers and found differences between cells with functional SETD2 and those without it.”
As they had found when studying cell migration, disrupting SETD2’s ability to methylate actin at lysine-68 caused defects in actin polymerization. In autophagy, disrupted actin polymerization altered actin’s interaction with another protein called WHAMM. As a result, the cells had autophagy defects. Significantly, there were no changes in the expression of any autophagy-related genes, further suggesting a role for SETD2 cytoskeleton modification rather than its chromatin function.
Find all the details of this work in the journal Biochemical and Biophysical Research Communications.
A rapidly growing field of cell biology
“Actin modifications, such as the addition of methyl groups described here, have been aptly dubbed the ‘Cinderella of the cytoskeleton’ and are only now being recognized as key regulators of cytoskeleton dynamics,” Seervai said. “Our findings and those of other groups are changing this perspective.”
More researchers are expressing interest in this new aspect of cytoskeleton regulation and we anticipate new discoveries pointing at potential new therapies for conditions involving cytoskeleton defects.”
Seervai was also involved in organizing a special interest subgroup at the ASCB/EMBO Cell Bio 2020 virtual meeting on post-translational modifications of the cytoskeleton, including actin and tubulin. “We had nearly 300 people, including the ASCB president, at our session this year. From what I’ve heard, this is a sign that things have turned a corner for the field since such a session was first organized several years ago.”
Other contributors to this work include Menuka Karki, Rahul Jangid, Durga Nand Tripathi, In-Young Park, Sandra Grimm, Cristian Coarfa, and Sung Yun Jung from Baylor College of Medicine; Sarah Kearns, Kristen Verhey, and Michael Cianfrocco from University of Michigan; Brian Millis and Matthew Tyska from Vanderbilt University; and Frank Mason and W. Kimryn Rathmell from Vanderbilt University Medical Center.
This work is supported by grants from the American Heart Association (19PRE34430069), the NIH (NCI-R35CA231993, R01CA203012, R35GM131744, P30ES023512, P30CA125123, NCI-CA125123, NIH-RR024574), the Templeton Foundation (no. 61099), CPRIT (RP170005, RP180672), the Department of Defense (KC170259) and the William and Ella Owens Medical Research Foundation.