Evolution meets biochemistry to better understand how dopamine receptors work
When proteins fail at doing their job, disease usually follows. To perform their job, proteins need to maintain a particular structure and specific motions, and how proteins achieve that is still not completely understood. In an article published in the Proceedings of the National Academy of Sciences, Baylor College of Medicine researchers have contributed a new piece to the puzzle that helps explain how proteins maintain their structure and function, and opens the possibility for better drug designs.
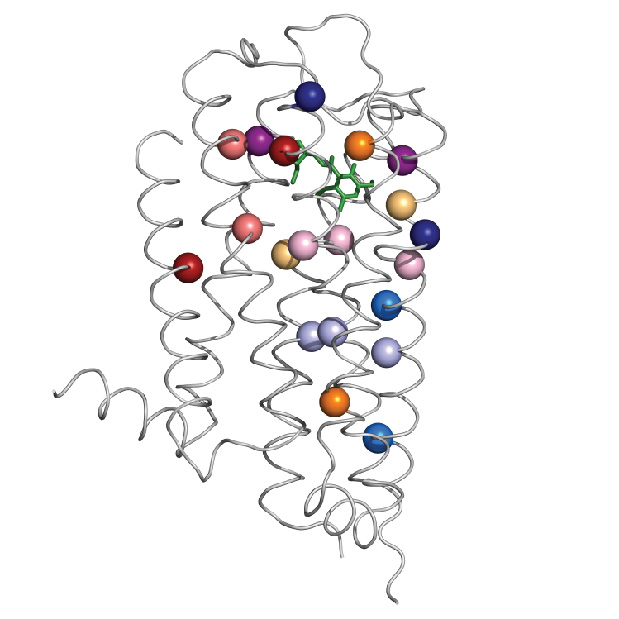
Dr. Olivier Lichtarge, professor of molecular and human genetics, the Verna and Marrs McLean department of biochemistry and molecular biology and pharmacology, as well as director the Computational and Integrative Biomedical Research Center at Baylor, and Dr. Theodore G. Wensel, professor and chair of the Verna and Marrs McLean department of biochemistry and molecular biology, professor of neuroscience, ophthalmology and pharmacology, led the team that developed a new mathematical tool that, together with biochemical analyses, allows them to pinpoint specific structural changes in the dopamine 2 receptor. These changes help maintain the receptor’s structure and function throughout an evolutionary time scale.
The specific structural changes refer to amino acids, the building blocks of proteins. Amino acids link to each other like beads on a necklace, then the ‘necklace’ folds naturally into a specific structure that results in a particular function. This three-dimensional arrangement of the linear sequence of amino acids results in pairs of amino acids that are far apart from each other in the linear sequence to end up near each other in the folded arrangement.
In some cases, specific pairs of amino acids that end up near each other in the structure contribute to maintaining that structure and function of the protein, but, in other cases, pairs that end up physically separated interact through an intervening network of other amino acids. The latter is called allosteric communication. Finding out which amino acids are far apart and yet connected to each other in this fashion to make proteins work is not an easy job.
Usually, the study of how proteins work has been a field of biochemists

“In terms of experimental science, traditionally biochemists like me have tried to get at the relationship between protein structure and function by looking at structures and making alterations and seeing what it does to function. For example, the experiments in this paper took a period of years to do. It was painstaking, labor intensive work,” said Wensel.
The new approach to finding allosteric pairs involves a mathematical tool, called ET-MIp, which is based on principles of evolution. Through evolution, the dopamine 2 receptor has maintained its function while varying to certain degree the sequence of amino acids. Allosteric pairs involved in maintaining the structure and function of the receptor have been conserved. The mathematical tool helps identify the pairs of amino acids most likely to be involved in allosteric communication.
By combining biochemistry work with ET-MIp, which incorporates computational analysis of many different sequences of dopamine 2 receptors and data of evolutionary divergence, the researchers were able to identify pairs of amino acids involved in allosteric communication.

“More often than not, pairs that we believed had an evolutionary signature for coupling [allosteric communication], ended up experimentally behaving as an allosteric pair. There seems to be a signal that we can find during evolution and experimentally the signal is validated,” said Lichtarge.
This discovery is significant because the dopamine 2 receptor, which is present in numerous regions of the central nervous system, has been implicated in neurological and psychiatric conditions, such as Parkinson’s disease, schizophrenia and depression. By contributing to a better understanding of how the function of the dopamine 2 receptor is regulated, the researchers present with potential new targets for better drugs.
Yun-Min Sung and Gustavo J. Rodríguez from the Verna and Marrs McLean department of biochemistry and molecular biology, and Angela D. Wilkins from the department of molecular and human genetics, all from Baylor, also contributed to this work.
This work was supported by NIH grants R01-GM066099, R01-EY011900, R01-EY007981, R01-GM079656, and T90-DK070109, by National Science Foundation grants DBI-1356569, and by the Welch Foundation grant, Q-0035.