The road to uncovering a novel mechanism for disposing of misfolded proteins
About 30 years ago, Dr. Richard Sifers set out on a journey to discover why people with a rare condition known as alpha1-antitrypsin (AAT) deficiency present with high variation in the severity of liver disease. His journey led him to the discovery of fundamental underpinnings of this condition and, unexpectedly, to uncovering a novel cellular mechanism for disposing of misfolded proteins. The latter has implications not just for AAT-deficiency, but also for other more common conditions associated with accumulation of defective proteins, including neurological disorders, such as Alzheimer’s disease.

AAT deficiency can develop in people who carry the AAT gene with a mutation called Z.
“I began studying AAT deficiency because I was intrigued by the wide range of severity of the condition. Some of the people carrying two copies of the Z mutation developed lung disease late in life and some developed liver disease. Interestingly, the condition also could appear very early in life. Some newborns and infants developed severe liver disease and needed to have a transplant to live,” said Sifers, professor of pathology & immunology and member of the Dan L Duncan Comprehensive Cancer Center at Baylor College of Medicine.
Other groups had shown that about 1 in 1,700 people carry two copies of the AAT-Z gene. However, only about 17 percent of these newborns with AAT-Z had clinically significant liver disease, and less than 3 percent of those progressed to life-threatening, end-stage disease as infants.
One of Sifers’s first contributions was to help develop the first screening test to determine whether a newborn was at risk of developing severe liver disease.
“Developing the screening method made me realize that I could tell whether a child was at a high risk of having liver disease, but still did not know what was causing the condition,” Sifers said.
Understanding AAT-deficiency
AAT is a protein produced by the liver and transported through the blood to the lungs, where it protects them from damage caused by other enzymes that breakdown proteins in the lung. The Z mutation produces a defective AAT protein that cannot fold into an appropriate 3-D conformation. Inappropriately folded AAT-Z proteins cannot exit the liver, so they do not travel to the lungs to protect them from destruction. This can lead to lung damage contributing to emphysema and other lung conditions.
“As I studied the disease, I noticed that AAT-Z, which should be released from the liver, was actually accumulating,” Sifers said. “This suggested that the naturally disposing mechanism of the cell might not be working.”
Sifers and others dug deeper into how cells dispose of misfolded proteins. They discovered that cells shuttle defective proteins from their place of synthesis, the endoplasmic reticulum (ER), to the cytosol, where they are degraded in a cellular structure called a proteasome. Key to this process is to tag the proteins for destruction.
We showed that removing certain sugars from proteins would flag them for degradation,” Sifers said.
“Specifically, we found that the human enzyme mannosidase Man1b1 acted like a quality-control factor that mediated the removal of the sugar mannose from misfolded AAT-Z proteins, promoting their degradation.”
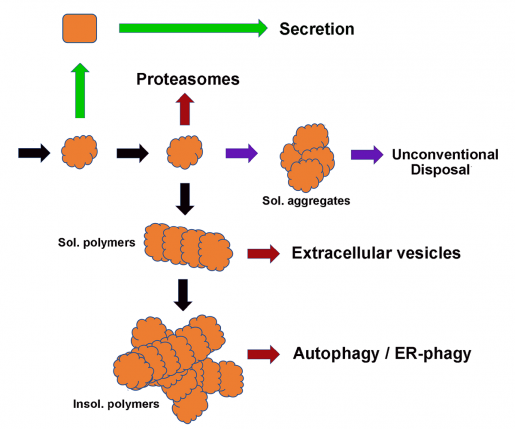
The AAT deficiency model has been used by many other researchers studying conditions also linked to toxic accumulation of misfolded proteins in cells, altogether called conformational diseases. This approach has accelerated the understanding of the underlying causes of these conditions, offering novel opportunities for potential treatments.
Connecting Man1b1 and AAT deficiency-associated liver disease in infants
Although researchers knew that liver injury associated with AAT deficiency was linked to accumulation of misfolded AAT-Z proteins in the liver, there was still no explanation for the severe liver disease in infants.
In a 2009 paper, Sifers and his colleagues studied liver tissue samples from unrelated infants or children older than 2 years who had undergone liver transplantation for end-stage liver disease. They also conducted genetic linkage and functional laboratory experiments with other cells cultured in the lab.
They showed that certain genetic modification, a single nucleotide polymorphism, that leads to changes in the expression of the Man1b1 gene results in lower levels of Man1b1protein in the endoplasmic reticulum of liver cells.
Sifers and colleagues proposed that lower levels of Man1b1 impair the liver’s capacity to deal with the accumulation of misfolded AAT-Z. This likely accelerates reaching the tolerable threshold for protein accumulation, resulting in earlier liver failure.
Having AAT-Z and a genetic variant that slows down the disposal of misfolded AAT-Z proteins can explain the condition in the youngest patients.
“I was delighted that after years of research, we had found an explanation for the mystery of AAT deficiency-associated liver disease in infants and wondered whether my lab would make other major contributions in the future,” Sifers said.
Man1b1 has more than one role

As Sifers and colleagues continued studying Man1b1, they unexpectedly came across a role for this protein that had not been described before.

“We found that, in addition to tagging misfolded proteins for degradation by enzymatically removing mannose groups, Man1b1 also promotes protein degradation by another mechanism that is independent from the first,” Sifers said.
Sifers and his co-authors, Dr. Ashlee H. Sun, now at Polypus-transfection Biotechnology, and Dr. John R. Collette, postdocs in his lab, reported in the Proceedings of the National Academy of Sciences, U.S.A., that the conventional enzymatic removal system resides in one side of Man1b1, the C-terminal domain. In contrast, the new unconventional system is controlled by the other side of Man1b1, the N-terminal domain. Further studies will elucidate whether and how both systems operate in synergy.
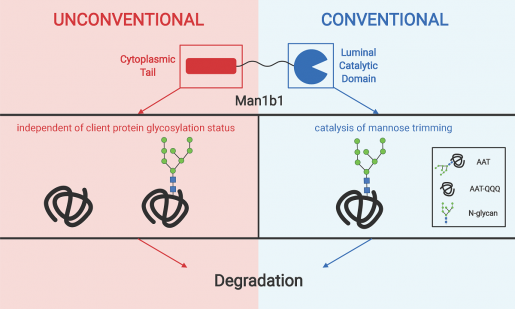
The researchers propose that the new unconventional system might be involved in the elimination of soluble protein aggregates that have been associated with conformational diseases. For instance, human Man1b1 has been linked to the causes of multiple congenital disorders of intellectual disability and HIV infection, and to poor prognosis in patients with bladder cancer.
“Our work is a clear example that studying rare diseases can bring solutions for more common conditions.”
By investigating a rare liver disease in babies, we have stumbled upon a pathway that could possibly be targeted to prevent more common neurological disorders occurring in late age,” Sifers said.
This study was supported by research grants from Alpha-1 Foundation.