How breast cancer can become resistant to hormone therapy
Despite the overall success of hormone therapy, breast cancer tumors in patients with metastatic disease often fail to respond. One new mechanism that can explain resistance to hormone therapy in breast cancer involves two significant changes in the cells – overexpression of the FOXA1 gene and increased production of IL-8.
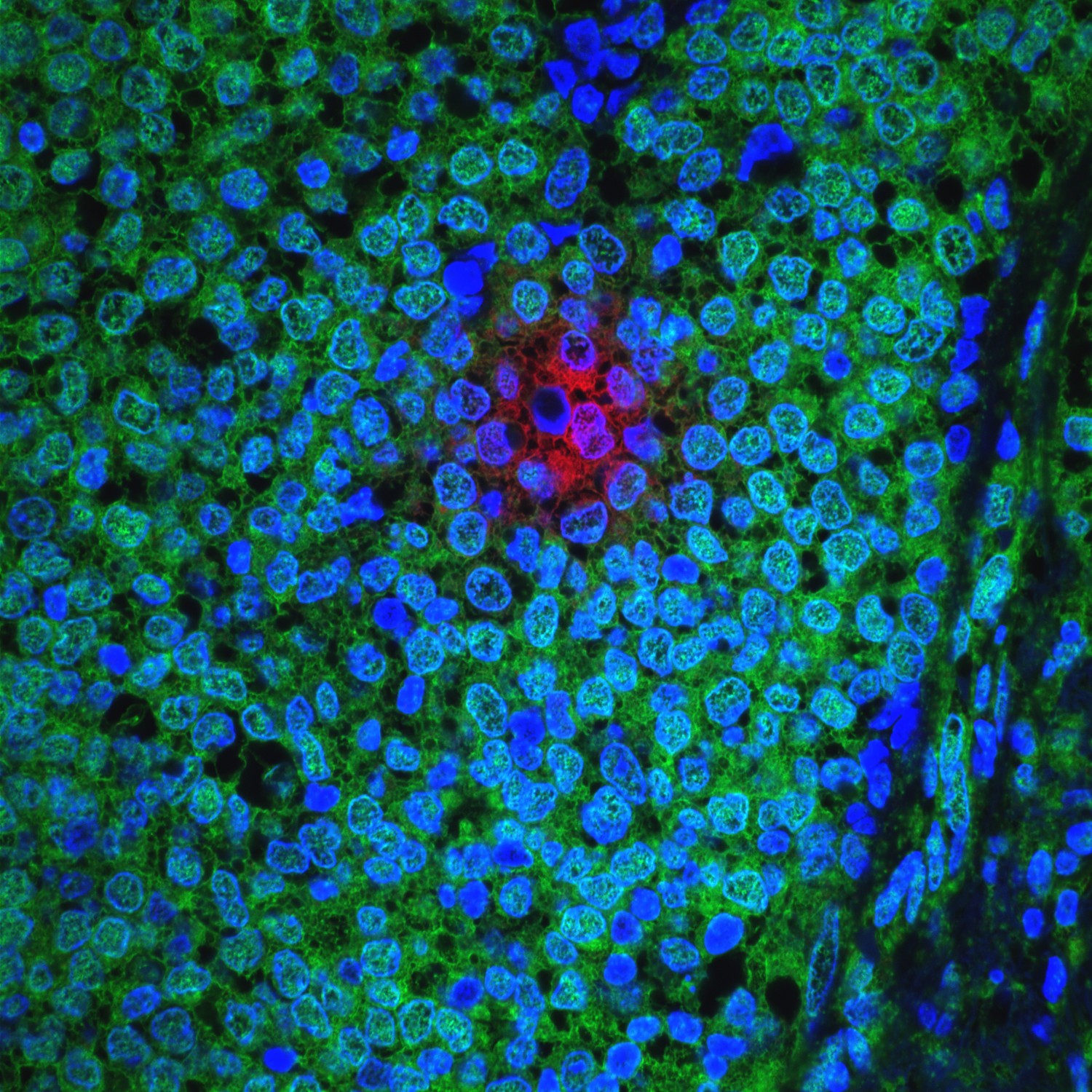
A cluster of breast cancer cells (red) within a human ER-positive primary breast tumor (cell nuclei in blue; rapidly cycling, AKT-high, cancer cells in green). (National Cancer Institute \ Dana-Farber Harvard Cancer Center at Massachusetts General Hospital)
About 75 percent of breast cancers have estrogen receptors, hence they are called estrogen receptor positive (ER-positive) breast cancers. Original ER-positive breast cancer cells depend on estrogen to grow, and therapies that made the estrogen unavailable to cells, called hormone therapies, can result in long-term remission in some patients. Tamoxifen, one of several types of hormone therapy, works by binding to and blocking the estrogen receptor on cancer cells.
However, most patients with metastatic disease whose tumors responded in the beginning to hormone therapy, eventually relapse and die due to acquired resistance to hormone therapy.

“Our goal in this work was to better understand how ER-positive breast cancer cells become resistant to tamoxifen hormone therapy,” said co-senior author Dr. Rachel Schiff, an associate professor in the Lester and Sue Smith Breast Center and in the departments of molecular and cellular biology and medicine at Baylor College of Medicine.
The results of this study can have implications in the clinical setting. “Understanding how tumors become resistant to this therapy will reveal new ways to treat them to circumvent and/or reverse this resistance, thereby improving patient survival,” said co-senior said co-senior author Dr. C. Kent Osborne, director of the Dan L Duncan Comprehensive Cancer Center and professor of medicine at Baylor.

Unraveling tumor resistance to hormone therapy
To study how cells become resistant to hormone therapy the scientists took a number of laboratory breast cancer cell lines that were susceptible to hormone therapy, called parental cells, and from each they developed cells lines that were resistant to tamoxifen hormone therapy.
“We sequenced the genes of our numerous cellular models of tamoxifen resistance and compared them with the genes of the corresponding parental cells,” said Schiff. “We hoped to find genetic differences between both groups of cells that would give us a hint of the causes of resistance. We found that cells resistant to tamoxifen, as well as to other hormone therapies, make more of FOXA1 protein than parental cells, which in some cases was due to the amplification of the FOXA1 gene.”
When the scientists knocked out the FOXA1 gene in tamoxifen-resistant cells, the cells became susceptible to tamoxifen therapy, confirming that the FOXA1 gene plays an important role in tamoxifen resistance.
The function of the FOXA1 gene is to promote the expression of many other genes. In this case, “we found that overexpression of FOXA1 results in the activation of a series of genes that increase the metastatic potential of cells, for instance, genes that favor cell migration and development of blood vessel,” said Schiff.
At the top of the list of genes activated by overexpression of FOXA1 is the gene for IL8, which also contributes to the survival of the cells.
To determine the relevance of these results in actual ER-positive breast tumor samples, the scientists analyzed samples from a tumor bank and found tumors with high levels of both FOAX1 and IL8 protein.

“We see a tremendous therapeutic potential in this study,” said first author Dr. Xiaoyong Fu, an assistant professor in Osborne-Schiff lab. “We can potentially design drugs directed not only to IL8 but also several of the genes activated by FOXA1 in tamoxifen-resistant cells to try to control the development of their metastatic potential.”
The research appears in the Proceedings of the National Academy of Sciences.
###
Other contributors to this work include Rinath Jeselsohn, Resel Pereira, Emporia F. Hollingsworth, Chad J. Creighton, Fugen Li, Martin Shea, Agostina Nardone, Carmine De Angelis, Laura M. Heiser, Pavana Anur, Nicholas Wang, Catherine S. Grasso, Paul Spellman, Obi L. Griffith, Anna Tsimelzon, Carolina Gutiérrez, Shixia Huang, Dean P. Edwards, Meghana V. Trivedi, Mothaffar F. Rimawi, Dolores López-Terrada, Susan G. Hilsenbeck, Joe W. Gray and Myles Brown. The scientists are affiliated with one or more of the following institutions: Baylor College of Medicine, Harvard Medical School, Oregon Health and Science University, Washington University and the University of Houston.
Financial support for this work was provided by the Breast Cancer Research Foundation, Stand Up to Cancer Translational Grant SU2C-AACR-DT0409, Susan G. Komen for the Cure Foundation Promise Grants PG12221410 and SAC110012, Department of Defense Breakthrough Award W81XWH-14-1-0326, NIH Breast Cancer Specialized Programs of Research Excellence Grants P50 CA058183 and CA186784, and NIH Cancer Center Grant P30CA125123.
This work was also supported by Proteomics and Metabolomics Core Facility with funding from CPRIT RP120092, and Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (P30AI036211, P30CA125123, and S10RR024574).