To be or not to be in the ER, that is the question
During the process of cellular protein synthesis mistakes can happen. Sometimes, proteins end up being misfolded. They do not shape up into the specific 3-D structure that is required for proper function. Misshaped secreted and transmembrane proteins usually trigger safety mechanisms that dispose of them by shuttling them from their place of synthesis, the endoplasmic reticulum (ER), to the cytosol, where they are degraded in a cellular structure called proteasome.
“Many proteins in the cell have sugar (glycan) chemical groups attached to them. Research has suggested that when these glycosylated proteins are misfolded, removing the N-glycan groups might be one of the steps required for their destruction,” said Dr. Hamed Jafar-Nejad, associate professor of molecular and human genetics at Baylor College of Medicine and lead scientist of the study.

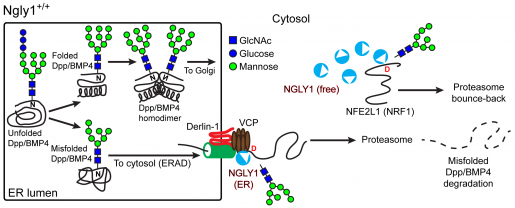
For many years, scientists have thought that once misfolded glycoproteins exit the ER and enter the cytosol, the enzyme N-glycanase 1 (NGLY1) removes their N-glycan groups, thereby facilitating the disposal of the misfolded glycoproteins by the proteasome.
“We found that for a critical signaling protein called BMP4, removal of N-glycans by NGLY1 does not occur after the misfolded molecules have been transferred into the cytosol, but is instead required for the transfer itself to be accomplished,” said first author Dr. Antonio Galeone, who was a postdoctoral fellow in Dr. Jafar-Nejad’s lab during the development of this project. He is currently in the Department of Biosciences at the University of Milan.
An intriguing finding
In humans, loss-of-function mutations in NGLY1 cause a multisystem developmental disorder called NGLY1 deficiency. Jafar-Nejad, Galeone and their colleagues from other institutions work with fruit fly and mouse models to investigate how NGLY1 mutations lead to developmental defects in various organs, hoping to find ways to treat this rare condition.
Previous findings from the Jafar-Nejad group had shown that the fruit fly equivalent of human NGLY1 is required to promote bone morphogenetic protein (BMP) signaling in a specific developmental context. However, how this actually happened at a molecular level remained a mystery. Moreover, whether mammalian NGLY1 plays a role in BMP signaling was not known.
In the current study, the researchers discovered that NGLY1 promotes the activity of one of the BMP pathway ligands called BMP4, both in fruit flies and mammals, by removing the N-glycan groups from misfolded BMP4 proteins.
But how does the degradation of misfolded BMP4 already removed from the ER contribute to BMP4 signaling mediated by properly folded BMP4 molecules that remain in the ER and are later secreted?
The researchers expected that the elimination of misfolded BMP4 proteins would happen as had been suggested for other proteins: defective BMP4 molecules would move into the cytosol, where NGLY1 removed the N-glycan groups, followed by proteasomal degradation.
Unexpectedly, they found that the N-glycan groups were removed before the defective BMP4 was fully moved from the ER into the cytosol. If the N-glycan groups were not removed, the defective BMP4 molecules did not transfer into the cytosol and accumulated in the ER.
Novel regulation of BMP4 signaling
Intrigued by these findings, Jafar-Nejad, Galeone and their colleagues reviewed the scientific literature and found previous work showing that NGLY1 is not exclusively a free cytosolic enzyme. Specifically, biochemical experiments had suggested that a small fraction of NGLY1 associates with the ER, although the functional significance of this association was not known.
The researchers showed that when misfolded BMP4 forms in the ER, NGLY1 is recruited to the ER through interaction with another protein called VCP.
Using laboratory-made NGLY1 mutations that impair NGLY1’s ability to bind VCP and be recruited to the ER without affecting its ability to remove N-glycan groups, the researchers showed that a perfectly functional NGLY1 that cannot be recruited to the ER cannot remove N-glycan groups from misfolded BMP4 molecules. This leads to the accumulation of misfolded BMP4 molecules in the ER and induction of ER stress, both of which may contribute to disease.

“Importantly, pharmacological inhibition of proteasomal function resulted in accumulation of de-glycosylated BMP4 in the cell, but did not impair BMP4 signaling, strongly suggesting that the critical function of NGLY1 in BMP4 signaling is to help remove misfolded BMP4 molecules from the ER,” Galeone said. “Once these molecules are in the cytosol, they do not inhibit normal BMP4 signaling anymore, whether they are degraded by the proteasome or not.”
These and other experiments led the researchers to propose that in normal conditions, accumulation of misfolded BMP4 in the ER triggers recruitment of NGLY1 to the ER. The ER-associated NGLY1 removes the N-glycan groups from the misfolded BMP4 molecules, promoting their transfer into the cytosol. This in turn allows properly folded BMP4 molecules to traffic from the ER to the extracellular space, where they will conduct their function.
A better understanding of NGLY1 deficiency
Before this study, NFE2L1 was the only biologically relevant, direct target of NGLY1 that had been identified in animals. NFE2L1 is critical in the activation of proteasomal gene expression and can only function when its N-glycans are removed by NGLY1.
The researchers’ findings identify a new critical target of NGLY1 and indicate that there is a division of labor in the function of NGLY1: only NGLY1 molecules recruited to the ER can remove glycan groups from BMP4, but NGLY1 molecules in the cytosol can remove glycans from NFE2L1, even when they are not recruited to the ER.
“This suggests that loss of NGLY1 not only leads to the accumulation of misfolded proteins in the cytosol, but can also result in the accumulation of other not-yet-identified NGLY1 targets in the ER,” Jafar-Nejad said.
The study also suggests that, in addition to mutations that inactivate NGLY1, mutations that affect NGLY1’s ability to be recruited to the ER might also cause some of the characteristics of NGLY1 deficiency observed in human patients.
Therefore, mutations that abolish NGLY1’s binding to VCP, but spare its enzymatic activity, might cause a yet-to-be-determined subset of the characteristics of NGLY1 deficiency observed in human patients.
“Identification of a new direct target of NGLY1 with broad roles in mammalian biology may help explain how NGLY1 deficiency affects multiple organs in human patients, and potentially guide the discovery of therapeutic approaches,” Galeone said.
BMP4 not only plays critical roles in animal development but also is implicated in certain cancers, such as ovarian and esophageal malignancies. Discovering important pathways involved in rare conditions such as NGLY1 deficiency also can benefit research on common diseases in which those pathways are involved.
Find all the details of this study in the journal eLife.
Other contributors to this work include Joshua M. Adams, Shinya Matsuda, Maximiliano F. Presa, Ashutosh Pandey, Seung Yeop Han, Yuriko Tachida, Hiroto Hirayama, Thomas Vaccari, Tadashi Suzuki, Cathleen Lutz, Markus Affolter and Aamir Zuberi. The authors are affiliated with one or more of the following institutions: Baylor College of Medicine, University of Basel, the Jackson laboratory, RIKEN Cluster for Pioneering Research, T-CiRA joint program and University of Milan.
This work was supported by grants from the Grace Science Foundation, the NIH (R35GM130317 to HJN) and H2020-MSCA individual fellowship (844147). Additional support was provided by Buzzati-Traverso fellowship, Fondazione AIRC per la Ricerca sul Cancro (20661), a postdoctoral fellowship from the Japan Society for the Promotion of Science, Research Fund Junior Researchers of the University of Basel, and Worldwide Cancer Research (18-0399). Imaging was performed at the Confocal Microscopy Core of the BCM IDDRC (U54HD083092; the Eunice Kennedy Shriver NICHD) and Unitech NOLIMITS microscopy facility core of University of Milan.