Preventing liver damage in animal models of Alagille syndrome
Alagille syndrome is a genetic disease that affects about 1 in 30,000 individuals. In most cases, the condition is caused by mutations in the gene JAG1, which affect multiple organs including the liver where it often results in cholestasis. In cholestasis, the flow of bile from the liver stops or slows, leading to bile buildup that in time causes liver damage. Current treatments focus on delaying disease progression; the only cure for liver disease in Alagille syndrome is liver transplantation.
“One of the major functions of the liver is to drain bile, which flows into the intestine to help absorb fats and eliminate toxins,” said corresponding author Dr. Hamed Jafar-Nejad, professor of molecular and human genetics at Baylor College of Medicine.

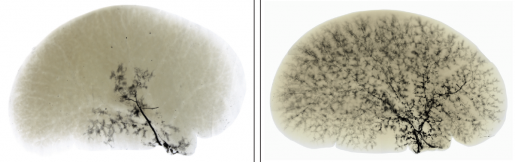
A healthy liver drains bile through a network of ducts called the biliary tree, but certain conditions such as Alagille syndrome, infections, toxins or injuries can reduce the number of ducts in the biliary tree, sometimes to less than half of what typically is present in a healthy liver. Fewer ducts, clinically known as bile duct paucity, mean less bile drains from the liver, which results in cholestasis that leads to liver disease.
Current treatments focus on correcting nutritional deficiencies, reducing bile accumulation and alleviating the severe itching caused by cholestasis to improve the quality of life and delay disease progression.
Developing a novel approach to prevent liver damage in Alagille syndrome
To establish a new approach to improve cholestasis in Alagille syndrome, the team worked with three animal models of the condition they had previously reported. The first model was genetically engineered to lack one of the two normal copies of the Jag1 gene (similar to Alagille syndrome patients) and had a moderate defect in the development of biliary trees. The other models were developed by manipulating a second gene in the livers of the first model, which resulted in severe to very severe defects in biliary tree development.
Interestingly, the Jafar-Nejad group had previously shown that genetically removing one copy of another gene called Poglut1 in their first Alagille syndrome mouse model significantly improved bile duct paucity. However, it was not known whether reducing Poglut1 gene activity after biliary defects had already occurred would still be beneficial in these models. In this study, the researchers investigated whether reducing POGLUT1 protein level after birth using a different approach would also improve the development of the biliary tree.
The team worked with Ionis Pharmaceuticals, a company that has pioneered the usage of antisense oligonucleotide (ASO) to treat human diseases. The Ionis team designed and generated an ASO specific for mouse Poglut1. ASOs are small stretches of modified DNA — sometimes containing RNA-like segments — that are commonly used to reduce the amount of a specific protein in the body (in this case, POGLUT1) by interfering with the process that produces the protein. ASOs are already in clinical use to treat some diseases but, to the researchers’ knowledge, no ASOs have been approved by the Food and Drug Administration to treat a biliary disease.
Encouraging results
“Our mouse models with moderate to profound biliary abnormalities received two postnatal injections of the ASO, which in turn reduced the activity of gene Poglut1 that produces the protein in the liver,” Jafar-Nejad said.
Importantly, the treatment significantly improved bile duct development and biliary tree formation and prevented fibrosis and cell death in the liver without adverse effects.”
Cell-based signaling assays indicated that reducing the level of protein POGLUT1 promoted an increase in the amount of JAG1 protein and JAG1-mediated signaling, which is likely to be the mechanism through which ASO improved the poorly developed biliary tree in all three animal models.
We are excited about these findings, as they show that it is possible to promote the formation of a near-normal biliary tree in animals that already suffer from significant bile duct paucity,” Jafar-Nejad said.

“Alagille syndrome is one of the most common genetic causes of cholestasis in children. Although much more work needs to be done to demonstrate the effectiveness of this approach to treat the human condition, our study offers for the first time a potential therapeutic option for these patients that bypasses the need for liver transplant. The thought that children suffering with this condition may find transplant-free relief keeps us motivated to work on the project,” said second author Duncan Fox, a graduate student in the Jafar-Nejad lab.

“Our study may not only be useful for treatment of Alagille syndrome, but it may also benefit patients with other conditions involving bile accumulation,” said first author Dr. Nima Niknejad, a postdoctoral associate in the Jafar-Nejad lab while he was working on this project and currently a senior scientist in Nitto BioPharma.
Find all the details of this work in journal Hepatology.
Other contributors to this work include Jennifer L. Burwinke, Neda Zarrin-Khameh, Soomin Cho, Armand Soriano, Ashley E. Cast, Mario F. Lopez, Kari A. Huppert, Frank Rigo, Stacey S. Huppert and Paymaan Jafar-Nejad. The authors are affiliated with one of the following institutions: Baylor College of Medicine, Cincinnati Children’s Hospital Medical Center or Ionis Pharmaceuticals.
This study was supported by the National Institutes of Health (grants R01 DK109982, R35 GM130317, R01 AR076770, R01 DK120765, R01 DK107553, R01DK132751, P30 DK56338), a Pilot/Feasibility Award from the Texas Medical Center Digestive Disease Center and two Alagille Syndrome Accelerator Awards from the Medical Foundation established by generous funding from an anonymous private donor. Further support was provided by the Department of Molecular & Human Genetics, Baylor College of Medicine, a T32 GM08307 training grant, and Ionis Pharmaceuticals.