Rap1 controls the body’s sugar levels from the brain, regulating it may help manage diabetes
Managing type 2 diabetes typically involves losing weight, exercise and medication, but new research by Dr. Makoto Fukuda and colleagues at Baylor College of Medicine and other institutions suggests that there may be other ways to control the condition through the brain. The researchers have discovered a mechanism in a small area of the brain that regulates whole-body glucose balance without affecting body weight, which suggests the possibility that modulating the mechanism might help keep blood sugar levels in a healthy range.
“A growing body of evidence strongly suggests that the brain is a promising yet unrealized therapeutic target for type 2 diabetes, as it has been shown that it can regulate glucose metabolism,” said Fukuda, assistant professor of pediatrics-nutrition at Baylor. “To further materialize this concept, it is of great interest to identify potentially druggable molecular targets mediating the brain’s antidiabetic effects.”

Regulating whole-body glucose balance from the brain
Research has shown that within the hypothalamic region of the brain, a small area known as the ventromedial nucleus of the hypothalamus (VMH) contains glucose-sensing neurons and regulates glucose metabolism in peripheral tissues.
“VMH neurons are thought to be crucial mediators of the neural glucoregulatory mechanism,” Fukuda said. “However, the signaling mechanisms within VMH neurons that mediate whole-body sugar control remain elusive. In this study, we identified a molecular pathway in the VMH that mediates whole-body glucose balance and involves Rap1, an enzyme known to mediate overnutrition-associated disorders.”
The researchers worked with a diabetes model of high-fat diet-induced obesity in mice, in which they either activated or eliminated Rap1 specifically in VMH neurons by using either genetic or pharmacological techniques.
They discovered that activation of Rap1 in the hypothalamus exaggerated the high blood sugar levels or hyperglycemia in the diet-induced obesity mouse model. In contrast, genetic loss of hypothalamic Rap1 decreased hyperglycemia in dietary obesity.
“Interestingly, the changes in glucose levels were observed without alterations in body weight, suggesting a primary role of Rap1 in glucoregulatory function,” Fukuda said.
Our findings that Rap1 activity can be regulated via pharmacological intervention provide proof-of-concept for the potential of targeting Rap1 signaling within the brain to improve glucose imbalance and induce antidiabetic effects.”
While having no effect on body weight regardless of sex, diet and age, Rap1 deficiency in VMH neurons markedly lowered blood glucose and insulin levels and improved glucose and insulin tolerance.
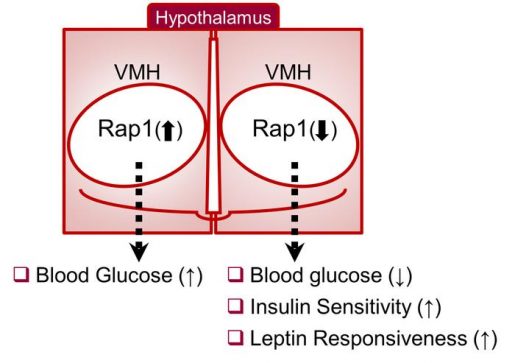
Taken together, the data suggest that hypothalamic Rap1 is a molecular pathway for the control of glucose metabolism and mediates high-fat diet-induced glucose imbalance, thereby making it a potential target for therapeutics.
“If we gain weight, blood glucose seems to be disturbed. That’s why obese people may have diabetes,” Fukuda said. “But in this mouse model we discovered that by modulating the activity of Rap1 in a small brain area we could regulate whole-body glucose metabolism without body weight change. There is still much work to do, but our findings suggest that maybe in the future obese people with diabetes could lower blood sugar levels by manipulating this mechanism of Rap1 in the brain without having to lose weight.”
Find all the details of this study in JCI Insight.
Other contributors to this work include Kentaro Kaneko, Hsiao-Yun Lin, Yukiko Fu, Pradip K. Saha, Ana B. De la Puente-Gomez, Yong Xu, Kousaku Ohinata, Peter Chen and Alexei Morozov. The authors are affiliated with one or more of the following institutions: Baylor College of Medicine, Kyoto University, Nagoya University, Cedars Sinai Medical Center, National Institute of Mental Health and Fralin Biomedical Research Institute at Virginia Tech Carilion.
Financial support for this project was provided by U.S. Department of Agriculture Current Research Information System 6250-51000-055, AHA-14BGIA20460080, NIH-P30-DK079638, NIH R01DK104901, and NIH R01DK126655; NIH awards R01DK093587 and R01DK101379; AHA-15POST22500012, the Uehara Memorial Foundation, Takeda Science Foundation and Japan Foundation for Applied Enzymology. Further support was provided by NIH awards R01MH120290, R01HL103868 and R01HL120947. This project was also supported in part by the Mouse Metabolic and Phenotyping Core at Baylor College of Medicine with funding from NIH UM1HG006348 and NIH R01DK114356.