Focused on the Heart — Novel mouse model of myotonic dystrophy displays reversible cardiac disease features of the condition
People with myotonic dystrophy type 1 (DM1), the most common adult-onset form of muscular dystrophy, progressively lose muscle mass and strength in their lower legs, hands, neck and face. The effects of the condition extend to the cardiac and central nervous systems and the gastrointestinal organs.
The laboratory of Dr. Thomas A. Cooper, professor of pathology and immunology, of molecular and cellular biology and of molecular physiology and biophysics at Baylor College of Medicine, has long been contributing to a better understanding of DM1. In his most current study, he and his colleagues focused on the cardiac aspects of the disease, which affects 50% of DM1 patients and is the second leading cause of mortality in individuals with the condition, after respiratory insufficiency resulting from skeletal muscle wasting.
“DM1 affects various aspects of cardiac function, but primarily involves conduction delays and arrhythmias, a problem with the electrical system of the heart,” Cooper said. “The condition also has been associated with cardiac fibrosis, fatty infiltration and other alterations, but how these changes happen at the molecular level is still not clear.”

What causes DM1
DM1 is caused by a mutation in the DMPK gene that results in a major expansion of a section of the gene formed by three-DNA building block repeats (CTG). While the unaffected population carries 5 to 37 repeats, people with the condition have 50 to 3000 repeats.
“When the mutated DMPK gene is transcribed into RNA, the RNA transcripts containing the repeat expansion accumulate in the cell nucleus,” said Cooper, who also is the S. Donald Greenberg and R. Clarence and Irene H. Fulbright Professor and a member of the Dan L Duncan Comprehensive Cancer Center at Baylor.
The accumulation of material in the cell nucleus disturbs the normal cellular processing and distribution of proteins, such as muscleblind-like (MBNL), and induces increased expression of others, such as CELF1. MBNL and CELF1 are RNA-binding proteins involved in alternative splicing or the processing of RNA. Scientists think that alterations in alternative splicing play a central role in the development of DM1.
To get new insight into how this process affects the heart specifically, Cooper and his colleagues set out to develop a mouse model of DM1 that focused on the heart and would allow researchers to study the molecular mechanisms that contribute to the cardiac features of the disease and to explore therapies.
A heart-focused and reversible model of DM1
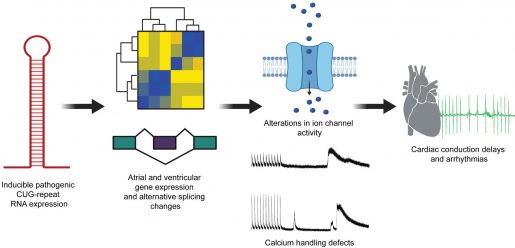
“We developed a mouse model in which only the heart cells would express RNA with 960 CUG repeats in the context of the human DMPK gene,” Cooper said. “We were quite pleased with the model when we observed that the animals reproduced many of the cardiac characteristics observed in the disease.”
Compared to the control animals, those carrying the repeats showed conduction problems, spontaneous arrhythmias and a higher predisposition of pacing-induced arrhythmias. Furthermore, mice with the repeats also showed accumulation of the CUG repeat containing RNA in cardiac cell nuclei, in which MBNL proteins also accumulated. The researchers also found strong changes in alternative splicing resulting in the reversion to fetal alternative splicing patterns, as it typically occurs in DM1.
Further analyses identified several gene expression and alternative splicing changes in genes associated with ion transport and calcium handling, which could be associated with alterations in cardiac function.
We designed our model to allow us to ‘turn down’ the expression of the RNA with the repeats,” Cooper said. “When we reduced it, the animals recovered their normal heart conduction, reduced RNA accumulation and colocalization of MBNL proteins and reversed the alternative splicing defects.”
Ours is the only heart model that has this length of repeats and presents strong similarities with the condition,” Cooper said.
“This model allows the field to focus on disease mechanisms in heart muscle, in addition to skeletal muscle where the focus has been until now. The goal with this model is to expand therapeutics to target the heart as well.”
For all the details about this study, go to the Journal of Clinical Investigation Insight.
Other contributors to this work include Ashish N. Rao, Hannah M. Campbell, Xiangnan Guan, Tarah A. Word and Xander H.T. Wehrens, all at Baylor College of Medicine, and Zheng Xia, who is at Oregon Health & Science University, Portland.
For a complete list of the sources of financial support for this project, go to the publication.