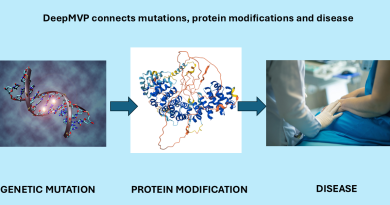
Aminode lets you do in less than a second what used to take a week
On his first year at Rice University, Kevin Chang, now a senior cognitive sciences major and ecology and evolutionary biology minor, knocked on many doors, including the one of the Sardiello lab at Baylor College of Medicine. The door opened giving Chang the opportunity to develop Aminode, a user-friendly, web-based tool researchers can use to gain insight on the function of proteins and their role in human diseases.
Chang spearheaded the development of Aminode after a flash of inspiration while working as part of his Rice undergraduate research requirement in Baylor College of Medicine. Along the way, he collaborated with co-author and now Rice alum Junyan Guo ’17 and Baylor postdoctoral associate Alberto di Ronza.
A flash of inspiration
Aminode helps researchers locate regions constrained by evolution in protein domains that have evolved across species and over time. Evolutionary contraints are factors that make populations resistant to evolutionary change.
“We’re trying to see which parts of the protein are the most conserved and do not change through evolutionary history,” Chang said. “The logic behind it is this: You have a human and you have a fish. Completely different organisms. Huge amount of evolutionary history behind them. Hundreds of speciation events. At each speciation event, there is potential for an amino acid to change.”
“So if you find the exact same amino acid at a certain index in the same protein in a human and fish, that means it’s probably really important,” Chang said. “If that amino acid changes in the human, it might cause a genetic disease.”
Aminode replaces the laborious process of comparing genetic code across multiple species from scratch, which requires using databases, bioinformatics tools and computer scripting. Aminode is a single platform that allows researchers to plug in a protein and see how it matches up across species for which code is already available.
“Instead of analyzing amino acids based on, say, 3-D structure or even multiple alignments, Aminode uses evolutionary relationships in many different species,” Chang said. “That makes it more relative and more functional.”
The idea for Aminode came to Chang shortly after he arrived at Rice and actively sought a part-time gig at Baylor. “I just went in and started talking to people,” he recalled. “I emailed a bunch of faculty members. I went to a lot of labs and one of them decided to take me on for a rotation. That was Marco Sardiello‘s.”
In Sardiello’s lab, Chang became fascinated by neurobiology and switched his Rice major from computer science to cognitive science with a specialty in neuroscience. He also encountered his first taste of genomic analysis.
“During my first rotation in the lab, a graduate student was looking at an analysis to justify studying a certain protein” to find mutations relevant to disease, Chang said. “That analysis was really big, and I said, ‘Hey, how’d you make this?’ He said, ‘Oh, man, it takes a long time,’ and he walked me through the procedure.”
“And I said, ‘I can make this a lot better.’ I think I said, ‘I can do this in two seconds.’”
![]()
 With Sardiello’s permission and the sponsorship of Michael Kohn, an associate professor in the Department of BioSciences at Rice, Chang built his first Aminode prototypes in the Python computer language and with Guo transferred it to the more sophisticated Java. Guo, now a software engineer at Microsoft, was sponsored on the project by Luay Nakhleh, a Rice professor of computer science and biosciences and chair of the Rice Department of Computer Science.
With Sardiello’s permission and the sponsorship of Michael Kohn, an associate professor in the Department of BioSciences at Rice, Chang built his first Aminode prototypes in the Python computer language and with Guo transferred it to the more sophisticated Java. Guo, now a software engineer at Microsoft, was sponsored on the project by Luay Nakhleh, a Rice professor of computer science and biosciences and chair of the Rice Department of Computer Science.
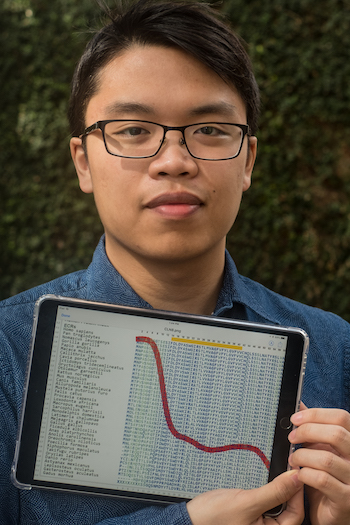
“Now we really can say that for a large gene, we can do in less than a second what used to take a week,” Chang said.

“I’ve never known a student like him,” Sardiello said of Chang. “He had a drive that was just amazing and worked literally night and day to get it done.”
Sardiello said his lab is using Aminode on several major projects to tag on proteins that can accept the minor modifications without the threat of making them pathogenic. “We need to have information about the best place in the protein to put this tag,” he said. “Aminode shows us the parts that are less likely to be altered by the addition.”
The lab also uses Aminode to find point mutations — single amino-acid alterations — to help locate the roots of genetic disease.
“Mutations in highly conserved regions are more likely to be pathogenic,” said Sardiello, whose lab will continue to develop the platform. “We know the pathogenic variations tend to fall in the regions of the protein that Aminode identifies as evolutionarily constrained.”
Kohn said evolutionary conservation data has not been used up to its potential for tracking mutations.
“What’s nice (about Aminode) is that (Chang) added phylogenetic information to it,” Kohn said. “His analysis knows that the chimpanzee and the macaque are more closely related to us than the dog. And it makes use of that information.”
Chang said he and Guo made the web interface as simple as possible for microbiologists, biochemists and other scientists who may not be familiar with evolutionary biology. “You don’t have to do anything other than know the gene name, type it in, press the button and get a high-quality analysis,” Chang said.
Aminode will tell you what parts of the protein are important, what parts aren’t important and where you can start an investigation,” Chang said. “If you’re a geneticist, you can see which parts of the protein you probably want to investigate for potential pathogenic mutations.”
“Obviously he got an A-plus on research,” Kohn said, pointing to the paper. “We’re getting a little more stingy on A-pluses because of grade inflation — but that is an A-plus if I’ve ever seen one.”
Read all the details of this work in the open-access paper in Nature’s Scientific Reports.
Courtesy of Mike Williams, senior media relations specialist in Rice University’s Office of Public Affairs.