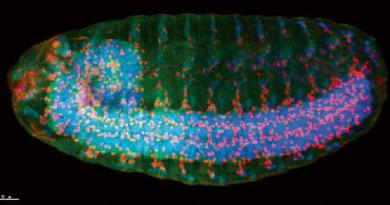
CRKL in 22q11.2; a key gene that contributes to common birth defects
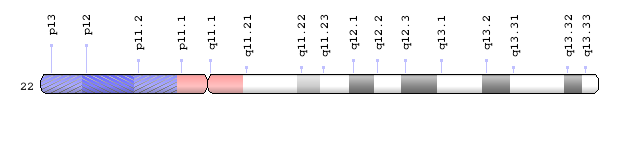
The 22q11.2 region of human chromosome 22 is a hotspot for a variety of birth defects. Scientists learned about this region because it is deleted in about 1 in 4,000 births, causing the loss of up to 40 genes. This chromosome microdeletion can result in a number of developmental abnormalities that vary greatly in severity among affected individuals. What many of the genes in this region do is not well understood, but when a set of these genes is absent it can cause havoc in the development and function of the heart, immune system and craniofacial features, as well as cognitive and behavioral issues. About 30 percent of individuals with the condition, called DiGeorge syndrome or 22q11.2 deletion syndrome, may also present with developmental abnormalities in the genitourinary system, both the upper- and the lower-tract defects.
Congenital genitourinary birth defects, whether they occur as part of a syndrome such as DiGeorge syndrome or as isolated congenital abnormalities, are among the most common types of birth defects.
Cryptorchidism, or undescended testis, occurs in about 6 percent of full-term male births, and hypospadias, a defect in which the opening of the urethra is not located at the tip of the penis, is seen in 1 in 250 male births. Defining the causes of genitourinary birth defects has been a focus of research investigations in Dr. Dolores Lamb‘s laboratory for many years.

“About 12 years ago, we began studying genitourinary birth defects with a technique called array comparative genomic hybridization, which is essentially like a molecular karyotype that has very high resolution so we can see little gains or losses in regions of chromosomes,” Lamb said. “We studied a number of unrelated children with cryptorchidism or hypospadias using this technology and found that about 20 percent of them had microdeletions or microduplications that clustered in specific regions of different chromosomes. One small deleted or duplicated chromosome region associated with these genitourinary conditions is 22q11.2. The children, however, were not diagnosed with DiGeorge syndrome.” The researchers found that the changes were ‘de novo,’ or new in the children, meaning they were not present in the parents.
Lamb and colleagues set out to identify which genes in 22q11.2 would be most likely involved in the abnormal development of the genitourinary system. If these genes were identified and their functions understood, researchers could then develop diagnostic tools and potential treatments for individuals affected by this condition.
Data from patients and animal models improve our understanding of genitourinary defects
Finding genes involved in developmental disorders is like finding the missing or altered pieces in a complex, broken machine for which we don’t have the blue print. Scientists use several strategies to find gene candidates and test their functions in the lab.
In this case, Lamb and colleagues took a two-pronged approach. On one side, they looked at copy number variations, both duplications and deletions, of genes in the 22q11.2 region of patients with DiGeorge syndrome who also presented with genitourinary abnormalities.
The analysis, together with creative thinking about potential pathways affected by a gene dosage change, led the team to suspect a gene called CRKL was the most likely candidate at 22q11.2 to be involved in genitourinary abnormalities as a result of gene duplication or deletion.
Further analysis showed that in humans CRKL is expressed in a variety of fetal tissues, including liver, lung, skeletal muscle, as well as in the heart, spleen, thymus, brain and kidney, which are relevant to DiGeorge syndrome. In the mouse and human, this gene is expressed modestly throughout development, including in the developing genitourinary tract. These results led the researchers to their next step toward determining CRKL’s involvement in genitourinary defects.
The researchers genetically engineered mice to lack crkl. One group of mice lacked both copies of the gene, the one received from the mother and the one passed on by the father, while another group lacked only one of the two crkl copies. Lacking both copies of the gene was lethal for the embryos, highlighting the importance of crkl in embryonic development. Analysis of both groups of embryos showed intrauterine growth restriction. In addition to having neural, heart and other congenital defects, about 23 percent of the mice exhibited severe kidney abnormalities. Like the human patients, the male mice lacking one copy of crkl had failure of testicular descent into the scrotum (cryptorchidism) resulting in fewer-than-average number of pups per litter, and with aging this sub-fertility progressed to male infertility. Further analysis showed that crkl regulates genitourinary development by altering expression of at least 52 DNA transcripts.

“Our data show that having CRKL gene dosage changes in this region, including the loss of one copy of CRKL, can negatively affect normal genitourinary (specifically testicular descent) and kidney development,” Lamb said. “CRKL has partial penetrance, so we see that some patients are affected while others aren’t. There is a spectrum of severity between different individuals and this inter-individual variation was present even in the mouse model.”
Implications for patients
“Our work has significant implications for initial patient diagnosis,” Lamb said. “The research findings imply that patients with genitourinary birth defects due to 22q11.2 changes in gene dosage should also be evaluated for other potential birth defects seen in patients with DiGeorge syndrome that would affect the patient’s future health.”
“This is important because some of the genes in region 22q11.2 affect brain development and behavior and/or cognitive function, autism spectrum disorder, schizophrenia, bipolar disorder, heart, hearing or autoimmune defects depending on which gene in this region is affected. The genitourinary birth defect may not be the only health issue needing to be clinically evaluated.”
Read all the details about this study in the Proceedings of the National Academy of Sciences.
Dr. Dolores Lamb is the director of the Center for Reproductive Medicine and a professor in the Scott Department of Urology and in the Department of Molecular and Cellular Biology at Baylor College of Medicine. She also is the Lester and Sue Smith Chair in Urologic Research.
Dr. Meade Haller (who undertook these studies as part of her doctoral dissertation) and Dr. Qianxing Mo, from Baylor College of Medicine, and Dr. Akira Imamoto from the University of Chicago, also contributed to this work.
This work was funded by National Institutes of Health Grants T32DK007763 and R01DK078121.
Would you like to learn more about research carried out in the Lamb lab? Visit this, and this links.