
Improving the view on the genetic causes of retinitis pigmentosa
Progressive development of night blindness and tunnel vision, sometimes from the early age of 2, are trademarks of retinitis pigmentosa. Being the most common inherited disorder of the retina, retinitis pigmentosa affects nearly 1 in 4,000 people. More than 1 million are visually impaired around the world due to this untreatable disease.
A number of genes have been associated with this condition. Fifty-eight are inherited in a Mendelian autosomal-recessive manner, while a number of genes are passed following an autosomal-dominant or X-linked mode of inheritance. However, these genes do not explain about 40 percent of the cases.

Doctorate student Smriti Agrawal, a co-first author of this study, and her mentor, co-senior author Dr. Rui Chen, associate professor of molecular and human genetics at Baylor College of Medicine, together with colleagues from other institutions in the U.K., California and Maryland have found that mutations in REEP6 — a gene that until now had not been associated with a human disease — can explain some of the cases that lacked a genetic diagnosis.
This discovery was the result of a long-term collaboration, which

began when Agrawal’s abstract at the renowned ARVO meeting of vision specialists caught the attention of researchers at University College London, who also study retinitis pigmentosa.
“We had discovered four families affected with this novel REEP6-associated retinitis pigmentosa in our patients and had begun preparing a manuscript when we read the abstract Rui Chen’s lab had sent to the 2016 ARVO meeting,” said co-senior author Dr. Michael Cheetham, professor of molecular and cell biology in the Institute of Ophthalmology at University College London. “When we learned that the Chen lab had a similar family and had developed mouse models with REEP6 mutations, we contacted Chen to discuss joining forces and collaborating.”

The collaboration allowed them to extend their search for genes in a larger patient database. The result is the identification of new mutations in REEP6 in seven patients from five independent families with retinitis pigmentosa.
A way to discovery: from the front to the back of the eye
“Both my grandparents have cataracts, a disease resulting from the crystalline lens, located in the front of the eye, becoming opaque,” Agrawal said. “This motivated me to study this condition, before joining Dr. Chen’s lab. Along the way, I became interested in other eyes diseases. In particular, I was very interested in studying the genetic basis of retinitis pigmentosa, a disease affecting the retina, a thin layer of tissue that lines the back of the eye. The retina is made of many different types of cells that communicate with each other to let us see. In retinitis pigmentosa, a type of light sensing neurons called the rod photoreceptors — required for low-light and peripheral vision— undergoes progressive degeneration, which eventually leads to severe vision impairment.”
“In this disease, the affected rods die early and cause the surrounding cone cells to die, eventually leading to blindness in the most severe cases,” said co-first author Dr. Gavin Arno, research associate in the Institute of Ophthalmology at University College London.”
Although the general characteristics of the different forms of retinitis pigmentosa are similar, they are genetically different. In addition, the manifestation of retinitis pigmentosa can vary significantly among patients. Some experience night blindness from the time they are 2 to 3 years old. However, the majority have symptoms when they are in their early teens; many don’t notice the progressive night blindness because they don’t drive, yet. The disease progresses to loss of peripheral vision, leading to tunnel vision. Sometimes, the end result is complete blindness. Other times, the patient may retain vision for life but still have decreased visual acuity.
“Understanding the genetics helps us project the natural path of the disease and, for instance, tell a teenager where his or her vision is likely going to be 20 to 30 years from now,” Chen said. “Once the rods die, they cannot be replaced, and there is no cure for the disease, yet. We hope that our research also will help us find ways to repair the damaged rods before it is too late.”
Bringing REEP6 to the table of retinitis pigmentosa research
The researchers analyzed all of the genes of nearly 600 patients with inherited retinal disease using next-generation sequencing. Seven of the patients with clinically diagnosed retinitis pigmentosa — all had tunnel vision and vision loss and were not related — had mutations in the same gene, REEP6.
“In all of the families studied, the disease was inherited in an autosomal-recessive manner,” said Agrawal. “Each of the parents does not present with the disease, but carries a defective copy of REEP6. The genetic mutations we identified in REEP6 differed among the patients.”
To further support the role of REEP6 in retinitis pigmentosa, the scientists carried out a series of experiments in which they overexpressed one of the REEP6 mutations in human cells lines. Then, they looked at whether the mutations affected the REEP6 protein — its stability, function, expression and localization.
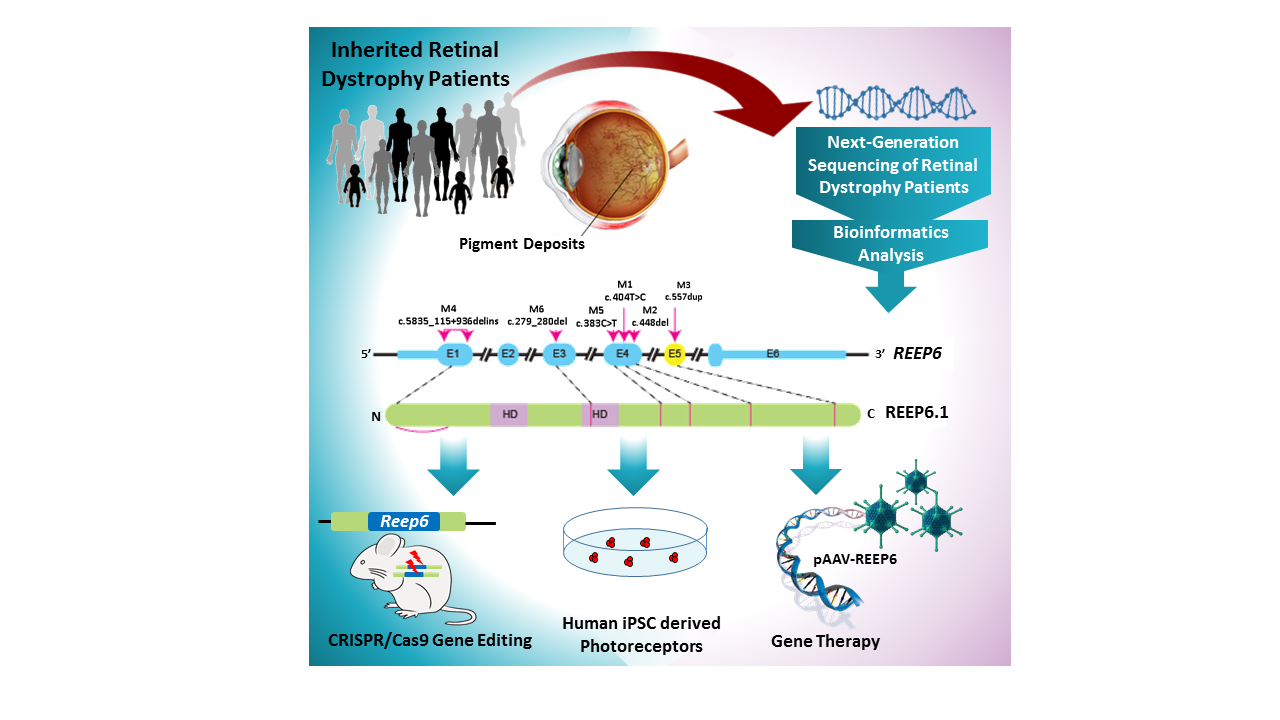
“We found that some of the variants cause formation of intracellular inclusion bodies as those seen in patient retinas, suggesting a level of protein instability” Chen said. “The mutant proteins do not fold properly and form aggregates that accumulate inside the cell as inclusion bodies.”
The scientists also developed a mouse model in which they studied the disease further.
“We selected one of the human REEP6 mutations and created the same specific point mutation in the mouse equivalent of REEP6 using CRISPR/Cas9 gene editing,” Agrawal said. “Mice carrying the mutation presented with characteristics equivalent to those found in patients. Our lab is the first to use CRISPR gene editing to create a patient-specific mutation mouse model of retinitis pigmentosa and using it to study disease pathogenesis.”
Opening new paths in retinitis pigmentosa research
“We are very excited about discovering a new gene associated with this disease,” Chen said. “It can potentially help diagnose retinitis pigmentosa, and the mouse model can help test gene therapies to rescue the defective gene as well as other therapies.”
“The new mechanism of disease we have discovered will allow us to study in greater depth how the retina works and maintains its function, and what happens when it goes wrong,” Cheetham said.
“I am incredibly thrilled to work on this project,” Agrawal said. “We found mutations in a new gene in patients with retinitis pigmentosa. I was amazed that the Reep6 mutant mice recapitulated the characteristics observed in the patients so well. Our work is exciting to me as it demonstrates the power of genetic tools and how we can use them to improve our knowledge of what causes blindness, a devastating condition that affects millions globally. I participated in a very productive international collaboration and I learned a lot along the way.”
“It has been a really positive and excellent collaboration between Baylor and UCL and we look forward to working together in the future,” Chen and Cheetham said.
The study appears in the American Journal of Human Genetics.
###
Other contributors to this work include Aiden Eblimit, James Bellingham, Mingchu Xu, Feng Wang, Christina Chakarova, David A. Parfitt, Amelia Lane, Thomas Burgoyne, Sarah Hull, Keren J. Carss, Alessia Fiorentino, Matthew J. Hayes, Peter M. Munro, Ralph Nicols, Nikolas Pontikos, Graham E. Holder, UKIRDC, Chinwe Asomugha, F. Lucy Raymond, Anthony T. Moore, Vincent Plagnol, Michel Michaelides, Alison J. Hardcastle, Yumei Li, Catherine Cukras and Andrew R. Webster.
The researchers are affiliated with one or more of the following institutions: University College London Institute of Ophthalmology, Moorfields Eye Hospital, Cambridge University Hospitals NHS Foundation Trust, University of Cambridge and University College London in the U.K.; as well as the University of California at San Francisco School of Medicine, the National Eye Institute at NIH and Baylor College of Medicine.
This project was funded by the Foundation Fighting Blindness (BRGE-0613-0618-BCM) and the National Eye Institute (R01EY022356, R01EY020540 and P30EY002520). The UKIRDC is funded by RP Fighting Blindness and Fight for Sight. This project was also supported by The Wellcome Trust (092621), The National Institute for Health Research (UK), Biomedical Research Center at Moorfields Eye Hospital and UCL Institute of Ophthalmology, Fight for Sight UK, Moorfields Eye Hospital Special Trustees, and Foundation Fighting Blindness – USA.

My husband has RP and we would like to see if anything can be done to help him. Is there a way to schedule an appointment with the Doctors at Baylor?? Any help would be greatly appreciated.
Good morning. You can request appointments here: https://www.bcm.edu/healthcare/request-an-appointment