Rhodopsin most likely works as a dimer in living cells
Although rhodopsin — the molecule that allows the eye to detect dim light — has a long and well-recognized history of more than 100 years, there is still controversy about the structure in which the molecule exists in the cells of the eye. A firm step forward toward better understanding rhodopsin structure in living cells shows for the first time that a dimer is the most likely configuration of rhodopsin in living organisms.
A team of scientists at Baylor College of Medicine, the University of Utah and the Johns Hopkins University School of Medicine have published their results in the Proceedings of the National Academy of Sciences, and hope this discovery will help develop future therapies for retinitis pigmentosa, a degenerative eye disease for which there is no known cure.
“Until recently, the established concept, based on a vast amount of literature from the fields of biochemistry, biophysics and physiology, sustained that rhodopsin exists and functions as a monomer — a single molecule. About 10 years ago, however, evidence began to emerge that rhodopsin may exist as a dimer, a two-molecule complex. Nonetheless, all of the supporting evidence available is from in vitro experiments, that is, experiments performed outside of a living organism,” said senior author Dr. Yingbin Fu, associate professor and Sarah Campbell Blaffer endowed chair of ophthalmology at Baylor. “In this study we have shown, for the first time in vivo — inside a living organism — that rhodopsin exists as a dimer.”

The structure and function of rhodopsin have long served as models for G-protein-coupled receptors (GPCRs), which constitute the largest family of proteins that allow the cell to sense the outside environment, including light, odors and hormones.
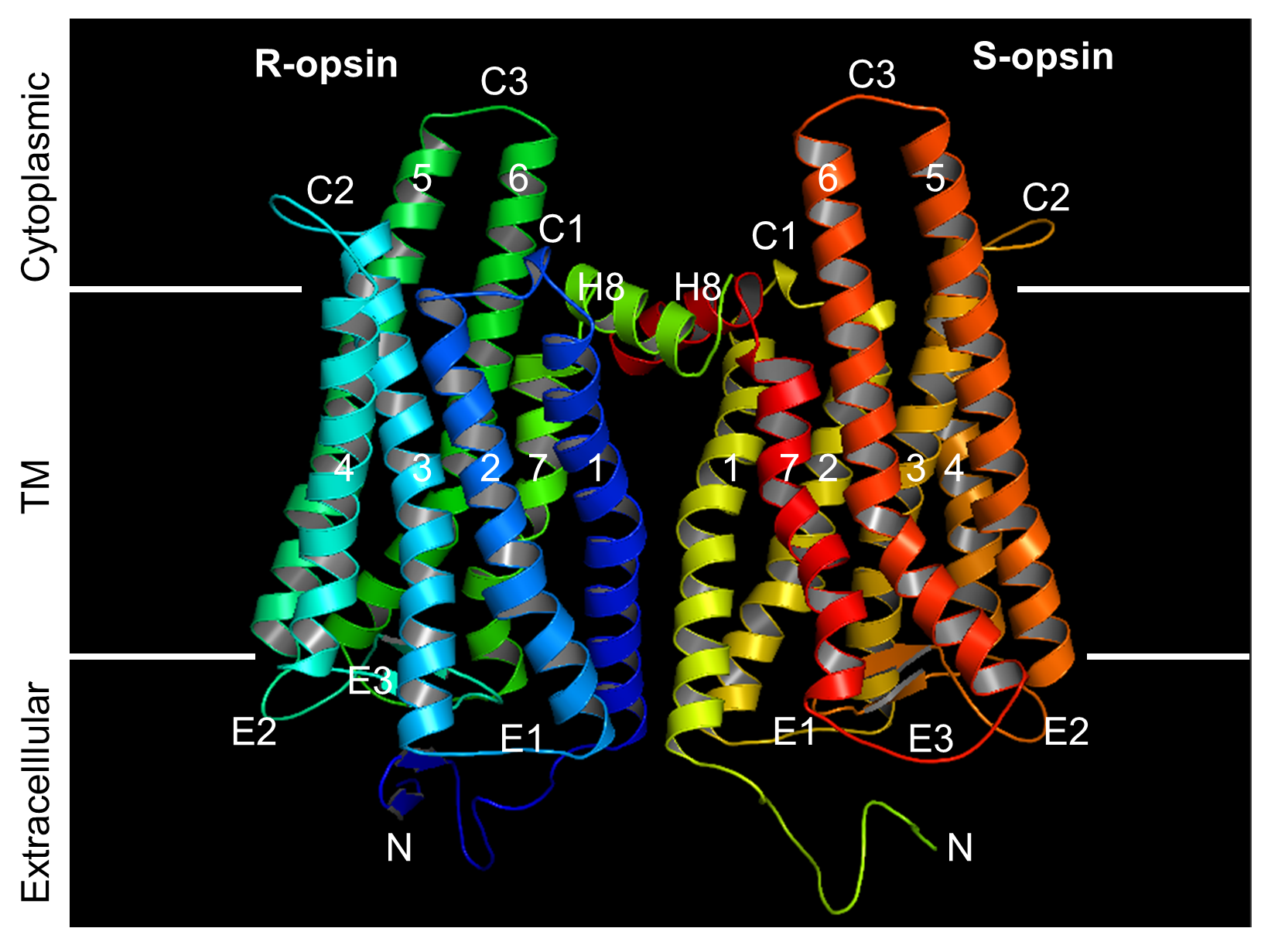
Many GPCRs have multiple subunits that are different from each other. Rhodopsin, on the other hand, has one subunit, thus making it difficult to distinguish rhodopsin monomers from rhodopsin dimers, in vivo. To determine whether rhodopsin forms monomers or dimers in vivo, the researchers tested rhodopsin’s ability to form dimers with a different molecule, a cone opsin.
“We obtained the first clear in vivo evidence by taking advantage of a unique genetically modified mouse line that expresses a cone opsin, a molecule responsible for color vision, in rods – the cells responsible for dim light detection that naturally express rhodopsin,” said Fu. “We found that in the absence of a vitamin-A based chromophore, a condition required for this experiment, the cone opsin can mature and target to the right place if and only if rhodopsin is present to help it along, that is, when cone opsin and rhodopsin form a two-molecule complex.”
Most importantly, the researchers also determined which sections or domains of the rhodopsin molecule were essential for forming dimers. “We have also confirmed a domain that is key for rhodopsin dimerization and shown, again in vivo, that rhodopsin maturation and targeting to its right place on the cell becomes defective when dimerization is disrupted by blocking the domains involved in forming dimers,” said Fu.
The authors hope that their findings will also help develop future treatments for retinitis pigmentosa, which has been associated with more than 100 mutations of rhodopsin.
“It is conceivable that some of the mutations that cause retinitis pigmentosa may affect the process of rhodopsin dimerization. When developing future therapies for this condition, it is important to keep in mind that rhodopsin works as a dimer,” said Fu.
“Considering that rhodopsin is the prototypical GPCR, that the evidence for a functional rhodopsin monomer is so strong, and that strong general resistance remains in the field against the rhodopsin-dimer concept, our in vivo experiments reported here are therefore crucial for steering the field in the correct direction,” said co-author Dr. King-Wai Yau, professor of neuroscience at Johns Hopkins University.

“As a clinician involved in multiple clinical trials of treatment for retinitis pigmentosa, I can say this is a very important finding,” said Dr. Timothy Stout, chair and professor of ophthalmology and director of the Cullen Eye Institute at Baylor. “We recruited Dr. Fu in September 2015 as a member of the new Center for Retinal Research at Baylor. This work is another demonstration of Dr. Fu’s innovative approaches and multidisciplinary expertise for studying retina diseases. I am confident that Dr. Fu’s team will continue to make great discoveries in retinal research.”
###
Other contributors to this work include Tao Zhang, Li-Hui Cao, Sandeep Kumar, Nduka Enemchukwu, Ning Zhang, Alyssia Lambert, Xuchen Zhao, Alex Jones, Shixian Wang, Emily Dennis, Amrita Fnu, Sam Ham and Jon Rainier from Baylor College of Medicine, the University of Utah and Johns Hopkins University School of Medicine.
This work was supported by the National Institutes of Health grants EY022614 and EY06837, NIH core grant 2P30EY002520, an unrestricted RPB grant to the Department of Ophthalmology at Baylor College of Medicine and the António Champalimaud Vision Award of Portugal.