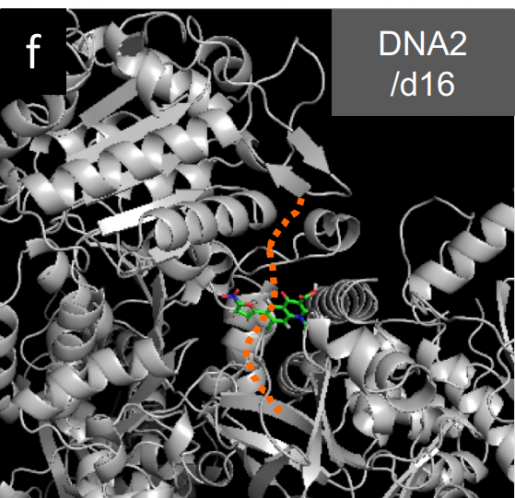
d16 reduces tumor growth and overcomes therapeutic resistance in mutant p53-bearing cancers
One of the most common alterations in many human cancers is mutations in p53, a gene that normally provides one of the most powerful shields against tumor growth. Mutations that alter the normal function of p53 can promote tumor growth, cancer progression and resistance to therapy, which are associated with poor prognosis. It is important to understand how p53 mutations help cancer grow to develop therapies to counteract their effects.
Studying how to target p53 mutations that promote cancer growth has been difficult. “One of the challenges has been to develop drugs that act on mutant p53 directly. Some of these drugs are under development, but they appear to be toxic,” said first author, Dr. Helena Folly-Kossi, a postdoctoral associate in Dr. Weei-Chin Lin’s lab at Baylor College of Medicine.

An indirect approach
For many years, the Lin lab approach has been not to interfere with p53 directly but to identify new vulnerabilities in cancer cells carrying p53 mutations that they could target to prevent or stop cancer growth. “In the current study, we focused on DNA2, an enzyme that binds to DNA and plays a role in its replication and repair. DNA2 is overproduced in cancer cells, particularly in those carrying a p53 mutation,” said Lin, professor of medicine–hematology and oncology and of molecular and cellular biology at Baylor. He also is a member of Baylor’s Dan L Duncan Comprehensive Cancer Center and the corresponding author of the work.

Research has shown that cancer cells with mutant gene p53 appear to be more dependent on DNA2 for survival than cells with normal p53. DNA2 stood out as a potential therapeutic candidate for mutant p53 cancers because DNA2 overexpression driven by mutant p53 gives cancer cells a survival advantage: it is associated with advanced disease, poor outcomes and acquired resistance to therapies.
“In the current study we investigated whether inhibiting the DNA2 function associated with the p53 mutation would stop cancer progression in cell-based and in animal models,” Folly-Kossi said.
The findings, published in the journal Cancer Research Communications, a journal of the American Association for Cancer Research, open opportunities for new combination therapies for these difficult-to-treat cancers.
Developing DNA2 inhibitors
Folly-Kossi, Lin and their colleagues searched for DNA2 inhibitors not by screening many compounds but with a computational approach that enabled them to identify the structures that are involved in DNA2 function. Then, they developed compounds that would fit into and bind to those structures, thus preventing DNA2 from binding to DNA and conducting its function.
Using this approach, we identified compound d16, which we tested in cell-based and in animal models and showed that it can suppress cancer growth,” Folly-Kossi said. “When we saw that d16 could reduce tumor growth when compared to placebo treatment, we started to believe that this could make a difference in someone’s life one day.”
The team also discovered that d16 can inhibit a DNA repair process and make cancer cells become sensitive to PARP inhibitors. PARP inhibitors are a class of FDA-approved drugs that only work in certain cancers with mutations in BRCA1 or BRCA2. Now, the combination of d16 with PARP inhibitors can work to treat many cancer cells harboring wild-type BRCA genes. They also found that treating with inhibitor d16 was able to overcome some conventional therapy resistance, making the cells susceptible again to other drugs that were ineffective before.
“We started with a concept, computer models and experimental data and then developed new drugs,” Lin said. “It was very satisfactory when we found that the inhibitor we had developed worked. It also confirms DNA2 as a good therapeutic target in mutant p53 cancers, a concept that we raised several years ago. Altogether, our findings validate our approach to fighting cancer, which we are continuing to use to find more new effective anticancer drugs.”
Other contributors include Joshua D. Graves, Lidija A. Wilhelms Garan and Fang-Tsyr Lin, all at Baylor College of Medicine. This work was supported by funding from NIH grant R01CA203824, Department of Defense Grants W81XWH-18-1-0329, W81XWH-19-1-0369, W81XWH-22-1-0226 and W81XWH-22-1-0534 and a Rivkin Center for Ovarian Cancer Pilot Award. Further support was provided by grants T32CA174647, T32DK060445 and T32GM136560.
Follow From the Labs on Twitter @BCMFromtheLabs.