Alzheimer’s disease risk genes alter neuroprotective mechanism
The brain has a natural protective mechanism against Alzheimer’s disease that is engaged when neurons face high levels of reactive oxygen species (ROS), natural byproducts of cellular metabolism linked to inflammation and other processes. The lab of Dr. Hugo Bellen has previously studied this mechanism and in this work the team worked with fruit fly and mammal models to further investigate whether known genetic risk factors for Alzheimer’s disease were associated with disturbing the protective mechanism, diving deep into the details of how this happened.
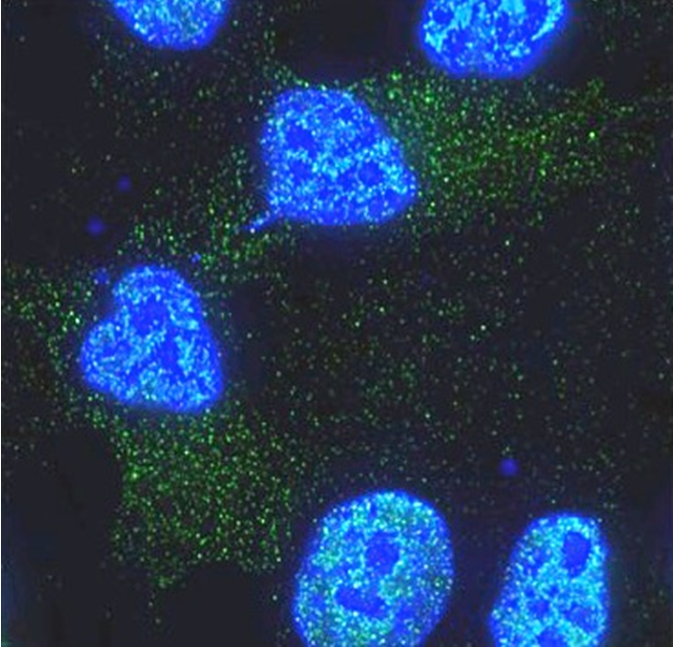
“Previous work conducted by Dr. Lucy Liu in the Bellen lab and colleagues showed that two brain cell types, neurons and glia, work together to protect against neurodegeneration,” said first author Dr. Matthew Moulton, a postdoctoral associate in the Bellen lab.

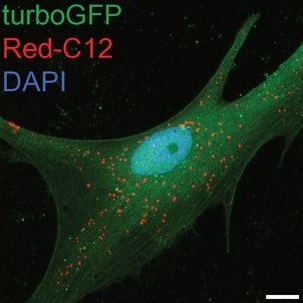
When ROS levels increase, which occurs with aging, different forms of stress or because of genetic factors, neurons produce abundant lipids. The combination of ROS and lipids results in peroxidated lipids, which deteriorate cellular health. Neurons try to avoid the damage by secreting these lipids, and apolipoproteins, proteins that transport lipids, carry them to glia cells. Glia store the lipids in lipid droplets, sequestering them from the environment, thus keeping them from damaging neurons.
In the previous work, the researchers connected the neuroprotective mechanism to the strongest genetic risk factor for Alzheimer’s disease, apolipoprotein APOE4. “We found that APOE4 is practically unable to transfer lipids to glia, while other two forms of APOE, APOE2 and APOE3, carry out the transfer effectively,” said Bellen, Distinguished Service Professor of molecular and human genetics at Baylor. “With APOE4, lipid droplet accumulation in glia is drastically reduced and the protective mechanism breaks down. This fundamental difference in the function in APOE4 likely primes an individual to be more susceptible to the damaging effects of ROS, which becomes elevated with age.”
In the current work, we wanted to identify genes that are critical for lipid droplet formation, specifically genes that are required for lipid export from neurons and lipid import into glia,” Moulton said.
“We looked at genes that interact with APOE in neurons to get the lipids out, and also in glia to get the lipids in. One reason we are interested in this comes from human studies that show that genes involved in both import and export of lipids have been implicated in Alzheimer’s disease and other related conditions.”
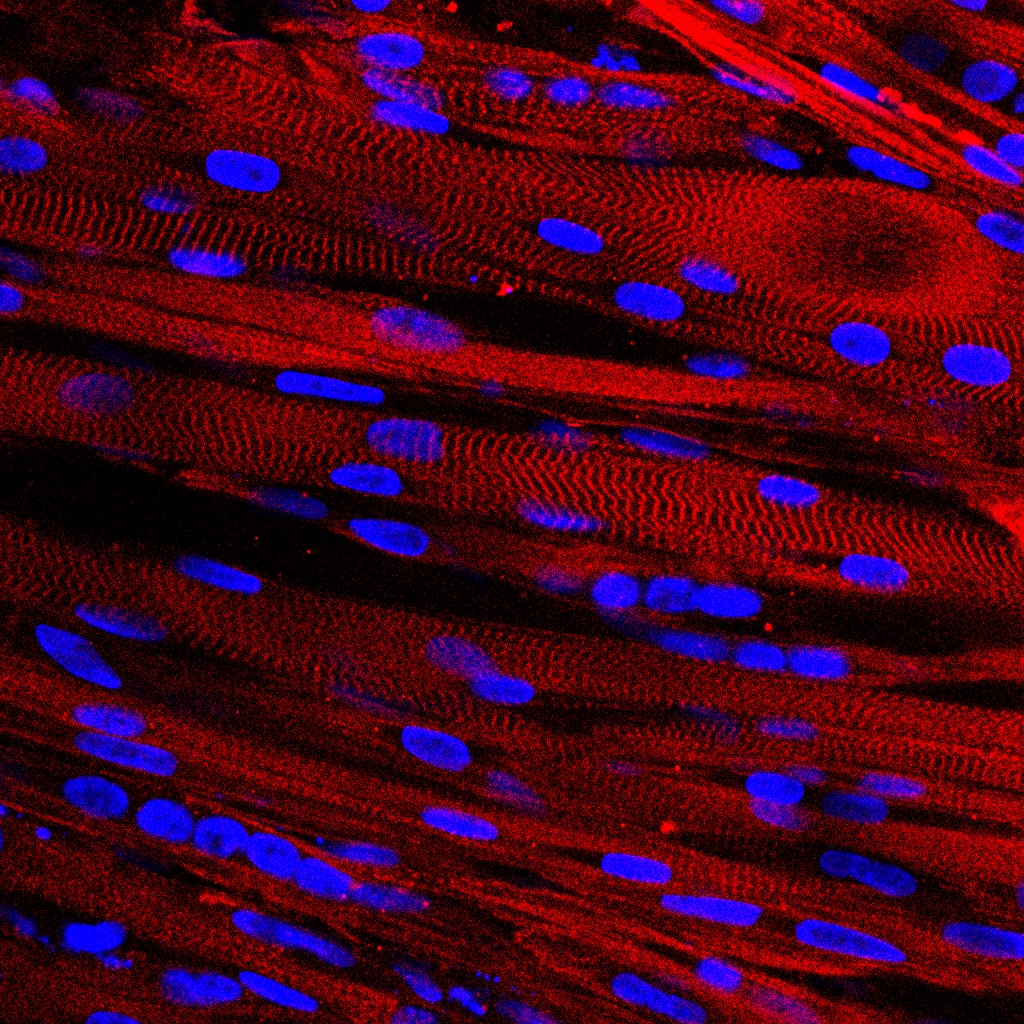
The team investigated the role of these Alzheimer’s risk genes in a fruit fly model, one gene at a time. The model allowed them to visualize, in the presence or absence of ROS, the effect of knocking down a particular gene, either in neurons or in glia, on the formation of lipid droplets, as well as on neurodegeneration.
“In all cases in which ROS was present and we saw droplet loss, we also saw neurodegeneration, again supporting that perturbations in glia droplet formation can lead to neuronal damage,” Moulton said.
With this approach, the team demonstrated that several genes that genome wide sequencing studies had associated with risk of developing Alzheimer’s disease disturbed neuroprotective lipid droplet formation, providing a mechanism that can explain the risk associated with these genes.

In addition, using the fruit fly model, Moulton and his colleagues tested whether an ABCA1 agonist, which was previously shown to restore APOE4’s ability to transfer lipids, could enable APOE4 to mediate lipid droplet formation in glia in the fruit fly model. “The ABCA1 agonist restored glial lipid droplet formation in an APOE4 fruit fly model, highlighting a potentially therapeutic avenue to prevent ROS-induced neurotoxicity,” said Bellen, Chair in Neurogenetics in the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital.
The researchers also investigated whether ROS could exacerbate the effect amyloid-beta may have on the disease. “We observed that ROS and amyloid-beta together increased neuronal death in fruit flies and resulted in larger and more numerous amyloid-beta-rich plaques in a mouse model, suggesting that, indeed, ROS and amyloid-beta can interact and potentially influence disease progression,” Moulton said.
As we age, ROS in the brain increases. If in addition there are mutations that disrupt the droplet pathways, then neurons can become sensitive to the accumulation of lipid droplets and this can pave the way to neurodegeneration,” Bellen said.
“Our findings support further investigations into feasible means to reduce the levels of ROS in the brain as a strategy to minimize ROS’s key contribution to neurodegeneration.”
Learn all the details of the study the Proceedings of the National Academy of Sciences.
Other contributors to this work include Scott Barish, Isha Ralhan, Jinlan Chang, Lindsey D. Goodman, Jake G. Harland, Paul C. Marcogliese, Jan O. Johansson and Maria S. Ioannou. The authors are affiliated with one or more of the following institutions: Baylor College of Medicine, Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, University of Alberta, Neuroscience and Mental Health Institute, Alberta and Artery Therapeutics, Inc.
Support for this work was provided by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health (NIH) award number P50HD103555, NIH award number U54HD083092, grants from the Texas Alzheimer’s Research and Care Consortium under award number 2018-497 05-11-II and from The National Institute on Aging of NIH, award number R01498 AG073260. Further support was provided by the Medical Genetics Research Fellowship Training Grant (NIH award number T32 GM07526-41), the Brain Disorders & Development Fellowship Training Grant (NIH award number T32501 NS043124-18), grants from Canadian Institutes of Health Research (MFE-64712, 173321) and the Heart & Stroke Foundation of Canada (award number 170722.) In addition, support was provided by Alberta Synergies in Alzheimer’s and Related Disorders (SynAD) program funded by the Alzheimer Society of Alberta and Northwest Territories through its Hope for Tomorrow program and the University Hospital Foundation, the Neuroscience and Mental Health Institute at the University of Alberta and the Howard Hughes Medical Institute.