Selecting the right deconvolution method just got easier
Biomedical scientists are increasingly using deconvolution methods, those used to computationally analyze the composition of complex mixtures of cells. One of their challenges is to select one method that is appropriate for their experimental conditions among nearly 50 available.
To help with method selection, Dr. Zhandong Liu and Haijing Jin, a graduate student in the Liu lab, have extensively evaluated 11 deconvolution methods that are based on RNA-sequencing (RNA-seq) data analysis, determining each method’s individual strengths and weaknesses in a variety of scenarios. From these analyses, Liu and Jin derived guidelines that scientists can use to determine the deconvolution method that optimally fits their needs.
“A great deal of work in biomedical research involves analyzing heterogeneous biological tissues to gain insight into the contribution of individual cells in, for instance, cancer growth or brain development,” said corresponding author Liu, associate professor of pediatrics-neurology at Baylor and director of the Bioinformatics Core of the Jan and Dan Duncan Neurological Research Institute.

Analyzing complex cellular mixtures is a difficult task. Researchers can conduct such analyses applying laboratory techniques that physically separate and/or identify cellular components, but these methods are time consuming and expensive.
Alternatively, researchers can use deconvolution methods that computationally extract information about individual cells in a complex mixture by analyzing large datasets derived from the bulk, such as RNA sequencing data.
For example, some researchers studying stem cells, a rare type of cell, might be interested in the percentage of these cells in the total blood cell population. They could conduct RNA-seq analysis of the bulk of cells and then apply a deconvolution method to determine the percentage of stem cells in the mixture. But, what method should they use?
In another example, if a scientist were interested only in the relative proportions of different cell types in a mixture, then one method would be best for deconvolution. But if the scientist wanted to find out the actual percentage of each cell type, then that deconvolution method would not be the best for that job, but another one that works better at providing that kind of answer. How can a scientist know which method works best in each situation?

“Our lab is one of many that developed deconvolution methods early on, contributing to the nearly 50 methods currently out there to do this type of job,” said first author Jin, a graduate student in Baylor’s graduate program of quantitative and computational biosciences. “The methods are based on different mathematical models and/or different assumptions to try to solve deconvolution problems, which involve basically how to go from a bulk heterogeneous tissue to profiles of individual cells.”
Because of this growing interest in deconvolution and the abundance of methods available, Liu and Jin felt that it was time to establish a guideline or benchmark to understand the strengths and the weaknesses of each method.
Running thousands of scenarios
The team studied 11 methods. They selected them according to the quality of the programing, the number of citations in the scientific literature and their popularity in the field.
“One of the challenges we faced was how to best test the strengths and weaknesses of each method in many possible scenarios,” Jin said.
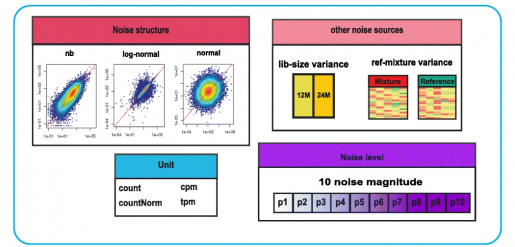
The researchers decided to use a computational or in silico approach that enabled them to simulate the thousands of scenarios necessary to test all the methods.
All these scenarios represented real-life experimental situations in cell research, cancer research or developmental biology. We simulated each one of them so we could identify the best deconvolution method for each scenario for people who are interested in applying these methods to their experiments,” Jin said.
“That’s the value of this work,” Liu said. “We are providing a benchmark study on various deconvolution methods and guidance for people working on different topics in biology to facilitate the analysis of their experimental results.”
Find all the details of this study in the journal Genome Biology.
This work was supported by National Institute of General Medical Sciences grant R01-GM120033, Cancer Prevention Research Institute of Texas grant RP170387, Houston Endowment, Chao Family Foundation and Huffington Foundation.