Novel mechanism connects SPEG, RyR2 and atrial fibrillation
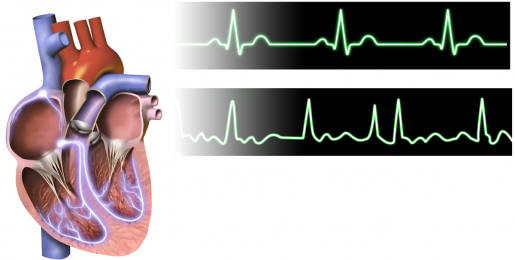
When the heart’s two upper chambers, the atria, beat out-of-sync with the two lower chambers, the ventricles, the result is atrial fibrillation (AFib), a serious condition that prevents the heart from pumping effectively. This uncoordinated heartbeat can lead to severe conditions, including heart failure, dementia and a fivefold increase in the risk of stroke. AFib affects more than 33.5 million people worldwide.

Dr. Xander Wehrens and his colleagues at Baylor College of Medicine’s Cardiovascular Research Institute and other institutions investigate what causes this type of arrhythmia looking to identify therapeutic targets to prevent this potentially deadly disorder.
“Past studies have recognized that enhanced diastolic calcium release via a type of calcium channel known as ryanodine receptors (RyR2) plays a role in AFib, but what triggers this enhanced release has not been fully explained,” said Wehrens, professor of molecular physiology and biophysics and director of the Cardiovascular Research Institute at Baylor.
Cells regulate the activity of calcium channels such as RyR2 by adding phosphate chemical groups to the channel with enzymes called protein kinases. In the current work, the researchers investigated the role of ‘striated muscle preferentially expressed protein kinase’ (SPEG), a novel regulator of RyR2 phosphorylation, in atrial fibrillation.
Connecting SPEG, RyR2 and atrial fibrillation
“When SPEG is functioning properly, it reduces RyR2 activity. So when we find reduced levels of SPEG, RyR2 is more hyperactive, and we see this abnormal activity in AFib,” said Wehrens, who holds the Juanita P. Quigley Endowed Chair in Cardiology.
This is one of the first examples of a kinase that has an inhibiting effect on calcium channels in the heart.”
Researchers first noted that patients with early state AFib had reduced levels of SPEG. To look further into this finding, they used a new atrial-specific gene therapy vector to selectively reduce SPEG levels in mouse models and found the mouse models to have an increased susceptibility to AFib.
AFib can progress from early-stage or paroxysmal AFib, meaning it comes and goes, to persistent AFib, which lasts until it is treated with medication or surgery, to persistent, long-standing AFib that doesn’t respond to treatments. Numerous factors can promote the development of AFib, including genetic variants, non-cardiac risk factors such as aging, obesity or alcohol abuse, and cardiac remodeling.

Our work suggests that modulating SPEG activity might represent a very specific target for the treatment and possibly prevention of AFib,” said Hannah Campbell, M.D./Ph.D. student in the Wehrens lab and first author on the study.
“The next step is to study this process in human tissue and to test whether enhancing SPEG activity could cure AFib in animal models.”
Read all the details of this work in the journal Circulation.
Others who took part in the research include Drs. Ann P. Quick, Issam Abu-Taha, David Y. Chiang, Carlos F. Kramm, Tarah A. Word, Sören Brandenburg, Mohit Hulsurkar, Katherina M. Alsina, Hui-Bin Liu, Brian Martin, Satadru K. Lahiri, Eleonora Corradini, Markus Kamler, Albert J.R. Heck, Stephan E. Lehnart, Dobromir Dobrev as well as researchers Brian Martin, Dennis Uhlenkamp and Oliver M. Moore. Affiliations include Baylor College of Medicine, University Duisburg-Essen, Essen, Germany, Brigham and Women’s Hospital, Harvard Medical School, University Medical Center Göttingen, Göttingen, Germany, Harbin Medical University, Harbin, China, and Utrecht University, Utrecht, The Netherlands. For full affiliate details see Circulation publication.
Funding is from the American Heart Association predoctoral fellowship 17CPRE33660059, and National Institutes of Health F30 fellowship HL140782. AQ was funded by AHA predoctoral fellowship 14PRE20490083, and NIH T32 training grant HL007676. TAW was funded by NIH T32 training grant HL139430. XW was funded through NIH grants HL089598, HL091947, HL117641, and HL147108. This work was performed during MH’s tenure as “The Kenneth M. Rosen Fellowship in Cardiac Pacing and Electrophysiology” Fellow of the Heart Rhythm Society supported by an unrestricted educational grant from Medtronic. BM was funded by NIH T32 training grant HL007676. SKL was funded through AHA postdoctoral fellowship 18POST34080154. SEL was funded by Deutsche Forschungsgemeinschaft through SFB 1002-S02, SFB 1190-P03, and IRTG-RP2. DD was funded by NIH grants R01-HL131517, R01-HL136389, and R01-HL089598 and the German Research Foundation (DFG) grant Do 769/4-1.