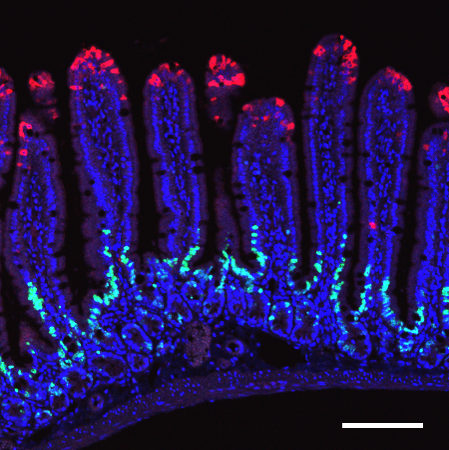
Losing neurofibromin is good news for breast cancer
It is well known that neurofibromin (NF1), a tumor suppressor produced by the NF1 gene, keeps cancer growth in check by repressing the activity of a cancer driver gene called Ras. It then follows that when NF1 is lost, Ras can drive cancer growth by promoting treatment resistance and metastasis. NF1, however, can do more than regulate Ras.
Drs. Eric C. Chang, Matthew Ellis and Zeyi Zheng at Baylor College of Medicine and their colleagues have discovered new insights into the function of neurofibromin that improve our understanding of breast cancer resistance and suggest novel therapeutic approaches to overcome it.
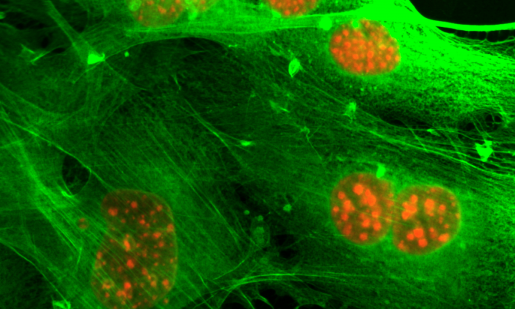
Neurofibromin loss drives tamoxifen and aromatase inhibitor resistance
The team first studied the importance of neurofibromin loss in a study they published in 2018. In this study, they sequenced tumor DNA seeking for mutations that can promote resistance to tamoxifen, a drug commonly used to prevent relapses from estrogen receptor positive (ER+) breast cancer.
“When we examined the mutational patterns in NF1, we observed that poor patient outcome only occurred when neurofibromin was lost, not through mutations that selectively affect Ras regulation. This suggested that neurofibromin may have more than one function,” said Chang, co-corresponding author of this work and associate professor in the Department of Molecular and Cellular Biology and a member in the Dan L Duncan Comprehensive Cancer Center’s Lester and Sue Smith Breast Center.


This thought triggered studies, spearheaded by Zheng in Chang’s lab, into the function of neurofibromin in ER+ breast cancer cells. One of his early experiments showed that when expression of NF1 is inhibited (to mimic neurofibromin loss in tumors), the resulting ER+ breast cancer cells were stimulated by tamoxifen instead of inhibited, as it usually happens. Furthermore, these neurofibromin-depleted cells became sensitive to a very low concentration of estradiol, a form of estrogen.
“The clinical relevance of these findings was immediately apparent because it suggested that tamoxifen or aromatase inhibitors, which lower estrogen levels available to the cancer cells, would be the wrong choice for treatment when neurofibromin is lost by the tumor,” said Ellis, co-corresponding author and professor and director of the Lester and Sue Smith Breast Center. Dr. Ellis also is a McNair Scholar at Baylor.
A key breakthrough: physically linking neurofibromin to estrogen receptor
Follow-up gene expression studies all strongly suggested that neurofibromin behaves like a classic ER co-repressor.
“A co-repressor must bind ER directly, but the group hesitated to conduct such an experiment without more evidence because it is not trivial to do so,” Chang said.
A breakthrough came when Dr. Charles Foulds, a co-author on the paper and assistant professor at the Center for Precision Environmental Health at Baylor, searched the “Epicome,” a massive proteomic database created by Dr. Anna Malovannaya and Dr. Jun Qin, also at Baylor. This is a part of an effort by Dr. Bert O’Malley, chancellor and professor of Baylor’s Department of Molecular and Cellular Biology to comprehensively document all the proteins associated with ER.

Foulds found neurofibromin in the database, which encouraged the team to ultimately investigate whether estrogen receptor and neurofibromin interacted directly. However, to seriously consider NF1 as an ER co-repressor, there was still another missing piece of the puzzle.
“One day Charles casually asked me whether neurofibromin had a region rich in the amino acids leucine and isoleucine, because co-repressors use these regions or motifs to bind ER, and it dawned on me that neurofibromin indeed does,” Chang said. “In fact, neurofibromin has two such motifs that mediate ER binding in a cooperative manner. These motifs are frequently mutated in cancer, but are not required for Ras regulation.”
Approaches to treat neurofibromin-deficient ER+ breast cancer
Since tamoxifen or aromatase inhibitors were found to be ineffective for neurofibromin-deficient ER+ breast cancer tumors, the researchers worked with animal models to determine whether the ER-degrading drug fulvestrant was still effective. However, fulvestrant only temporarily inhibited tumor growth because secondary Ras-dependent fulvestrant resistance was induced by neurofibromin loss. This Ras-dependent growth phase could be inhibited with the addition of a MEK inhibitor, which shuts off a key signaling pathway downstream of Ras.
The team validated this combination treatment strategy using a patient-derived xenograft (PDX) mouse model. In this model, a section of a human tumor taken from a patient is directly transplanted into a mouse under conditions that maintain the genomics and drug response of the original human tumor from which it was derived (Cell Reports, 2013). In this case, this PDX was derived from a patient who failed several lines of endocrine therapy and had already developed fulvestrant resistance.
The results of the combination of fulvestrant to degrade ER and a MEK inhibitor (e.g., selumetinib or binimetinib) to inhibit Ras downstream signaling, were encouraging — the tumor shrunk to almost undetectable levels,” Chang said.
“Our next goal is to test this combination therapy in clinical trials in order to determine its therapeutic potential in the clinic.”

“Neurofibromin is lost in at least 10 percent of metastatic ER+ tumors. As a result of these new data, we are now working on a clinical trial that combines a MEK inhibitor with fulvestrant,” said Ellis, Susan G. Komen scholar and associate director of Precision Medicine at the Dan L Duncan Comprehensive Cancer Center at Baylor. “Interestingly, MEK inhibitors are also being used to control peripheral nerve tumors in patients with neurofibromatosis, where a damaged NF1 gene is inherited. Our findings contribute to an understanding of why female neurofibromatosis patients also have a much higher incidence of breast cancer.”
Other contributors to this work include Meenakshi Anurag, Jonathan T. Lei, Jin Cao, Purba Singh, Jianheng Peng, Hilda Kennedy, Nhu-Chau Nguyen, Yue Chen, Philip Lavere, Jing Li, Xin-Hui Du, Burcu Cakar, Wei Song, Beom-Jun Kim, Jiejun Shi, Sinem Seker, Doug W. Chan, Guo-Qiang Zhao, Xi Chen, Kimberly C. Banks, Richard B. Lanman, Maryam Nemati Shafaee, Xiang H.-F. Zhang, Suhas Vasaikar, Bing Zhang, Susan G. Hilsenbeck, Wei Li and Charles E. Foulds. The authors are affiliated with one or more of the following institutions: Baylor College of Medicine, Chongqing Medical University, Adrienne Helis Malvin Medical Research Foundation, Zhengzhou University and Guardant Health.
This work appears in Cancer Cell,
See the publication for a complete list of the sources of support for this work.