Stopping antibiotic resistance before it happens
Antibiotic resistance is one of the most urgent threats to public health. One way bacteria develop resistance is by generating new mutations in their DNA that allow them to overcome the deadly effects of the antibiotic. In her lab at Baylor College of Medicine, Dr. Susan M. Rosenberg has been investigating this topic for many years and recently she and her colleagues have untangled the molecular events that allow bacteria to survive antibiotics.
“Traditionally, antibiotic resistance has been countered with new antibiotics. In this study, however, we looked to discover and then inhibit the molecular mechanisms that induce mutations conferring antibiotic resistance,” said Rosenberg, Ben F. Love Chair in Cancer Research and professor of molecular and human genetics, of biochemistry and molecular biology and of molecular virology and microbiology at Baylor.
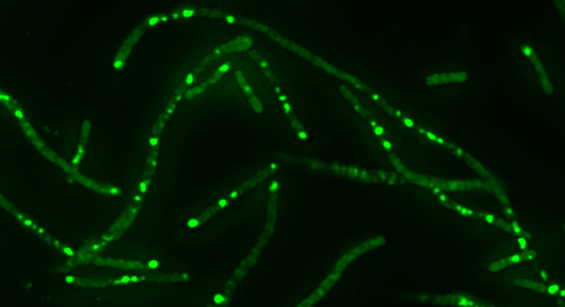
“We treated laboratory cultures of the bacterium Escherichia coli with low doses of the antibiotic ciprofloxacin, which are known to induce new mutations that confer antibiotic resistance,” said first author John P. Pribis, a student in the Integrative Molecular and Biomedical Sciences graduate program at Baylor and working in the Rosenberg lab. “In this system, we investigated in great detail the molecular pathways that were activated as the bacteria evolved antibiotic resistance.”
Pribis, Rosenberg and their colleagues discovered that ciprofloxacin induced DNA breaks, which were followed by bacteria releasing a burst of toxic compounds called reactive oxygen species. These compounds activated a general stress response that triggered error-prone DNA-repair, which resulted in mutations.
“We were surprised to find that the burst of reactive oxygen occurs only in 10 to 25 percent of the bacterial population, and that only these cells follow the next molecular steps that conclude in gene mutations,” Pribis said. “Some of these mutations may confer antibiotic resistance, but others will not and may be lethal for the bacteria. While these mutating ‘gambler’ cells sort of roll the dice on their genome generating mutations, the remaining bacteria do not mutate.”

“These findings suggest that bacteria that are exposed to low doses of antibiotic set in play a molecular strategy that confers a survival advantage to the bacterial population as a whole and is likely disadvantageous for some of the bacteria in an infection,” said Rosenberg, who also is leader of the Mechanisms in Cancer Evolution Program at the Dan L Duncan Comprehensive Cancer Center at Baylor. “While a small segment of the population, the gamblers, takes the risk of generating mutations, the rest of the bacteria stay stable and do not follow that path. As a result, if those that mutated do not survive, there still is a portion of the original population that will.”
The researchers’ detailed study of the molecular pathways leading to antibiotic resistance provided new opportunities to slow down the process, therefore reducing the chance of bacteria developing resistance. Pribis, Rosenberg and their colleagues looked for drugs that could slow down the process.
“We realized that there are FDA-approved drugs that reduce the production of reactive oxygen species, one of the crucial steps leading to mutations,” Pribis said. “We found that the drug edaravone, which is used for treating amyotrophic lateral sclerosis and stroke, would fit in the molecular mechanisms we had described inhibiting the production of reactive oxygen species.”
Indeed, we found that edaravone slowed down the development of mutations in bacterial cultures without losing its ability to kill bacteria. The drug prevented the rapidly evolving ‘gambler’ cell subpopulation from forming,” Rosenberg said.
The researchers plan to investigate next whether this drug is able to slow the course of an infection, with or without antibiotic treatment, in models of infections.
Read all the details of this work in the journal Molecular Cell.
Other contributors to this work include Libertad García-Villada, Yin Zhai, Ohad Lewin-Epstein, Anthony Wang, Jingjing Liu, Jun Xia, Qian Mei, Devon M. Fitzgerald, Julia Bos, Robert Austin, Christophe Herman, David Bates, Lilach Hadany and P.J. Hastings. For a complete list of author affiliations, click here.
This work was supported by NIH Grants R35-GM122598, R01-GM088653, R01-GM102679, R01-GM106373, T32-GM008231, Israeli Science Fund ISF 1568/13 and the Baylor College of Medicine (BCM) Integrated Microscopy Core funded by NIH (DK56338). Further support was provided by NIH: CA125123, the Dan L Duncan Comprehensive Cancer Center, postdoctoral fellowships RP160283 and 132206-PF-18-035-01-DMC (DMF) from the Cancer Prevention and Research Institute of Texas BCM Cancer Training Program and the American Cancer Society; the John S. Dunn Gulf Coast Consortium for Chemical Genomics and the BCM Cytometry and Cell Sorting Core (NIH: P30 AI036211, P30 CA125123, and S10 RR024574).