Crystal structure of PKGI suggests a new activation mechanism
By Ana María Rodríguez, Ph.D.
Kinases form a large family of proteins that seem to be involved in nearly every aspect of cell life. Faulty protein kinases have been found in a number of human conditions, including cardiovascular diseases, cancer and diabetes, and this has triggered much interest in understanding their regulatory mechanisms in order to design effective therapies.
Some protein kinases have similar structures, which has led some researchers to propose that the activity of these kinases is probably regulated in a similar manner and, therefore, may be targeted with similar medications. In a paper published in the journal Structure, a multidisciplinary team from Baylor College of Medicine, the University of Kassel, the King Abdullah University of Science and Technology, the Lawrence Berkeley National Laboratory and the University of California, San Diego have discovered that this may not always be the case.
“Kinases add a phosphate group to other proteins and this addition is like flipping a switch; it modifies the activity of the protein,” said Dr. Choel Kim, assistant professor in pharmacology and the Verna and Marrs McLean Department of Biochemistry and Molecular Biology at Baylor, and senior author of this report. “About 2 percent, or nearly 500, of all human genes are dedicated to coding protein kinases and over 50 percent of kinases are linked to various human diseases.”
Kim and colleagues are interested in studying the activation mechanism of protein kinase G I (PKG I). Their interest derives from their previous study published in The American Journal of Human Genetics in which they found a mutation of PKG I to cause thoracic aortic disease.
Understanding how PKG I regulates its activity
PKG I is a large molecule. It has a regulatory (R) domain and a catalytic (C) domain. When R and C domains are together, PKG I is inactive or dormant. It does not phosphorylate other proteins. To be active, PKG I needs a small compound called cGMP, a cyclic nucleotide, to bind to the R domain, which will then free the C domain to carry on phosphorylation of other proteins.
We think that targeting the R domain might be a way to effectively regulate the activity of PKG I involved in disease because this R domain is unique for this kinase. But first we need to understand how the R domain works,” said Kim.
The regulation mechanisms of both PKG and its closest homologue PKA are dependent on the high affinity interaction between the R- and C-domains in the absence of cyclic nucleotide and the drastic reduction of their affinity in the presence of nucleotide. In PKA, because the R- and C-subunits are separate polypeptide chains, they diffuse away upon activation, preventing possible reassociation that may causes unnecessary inhibition. Unlike PKA, the presence of the R- and C-domains on the same peptide chain in PKG can enhance the R-C interaction and this may cause this unnecessary inhibition even in the presence of cGMP. However, little is known about how PKG circumvents this unintended inhibition.
Crystal structure of PKG1 in its active form reveals a new regulatory mechanism

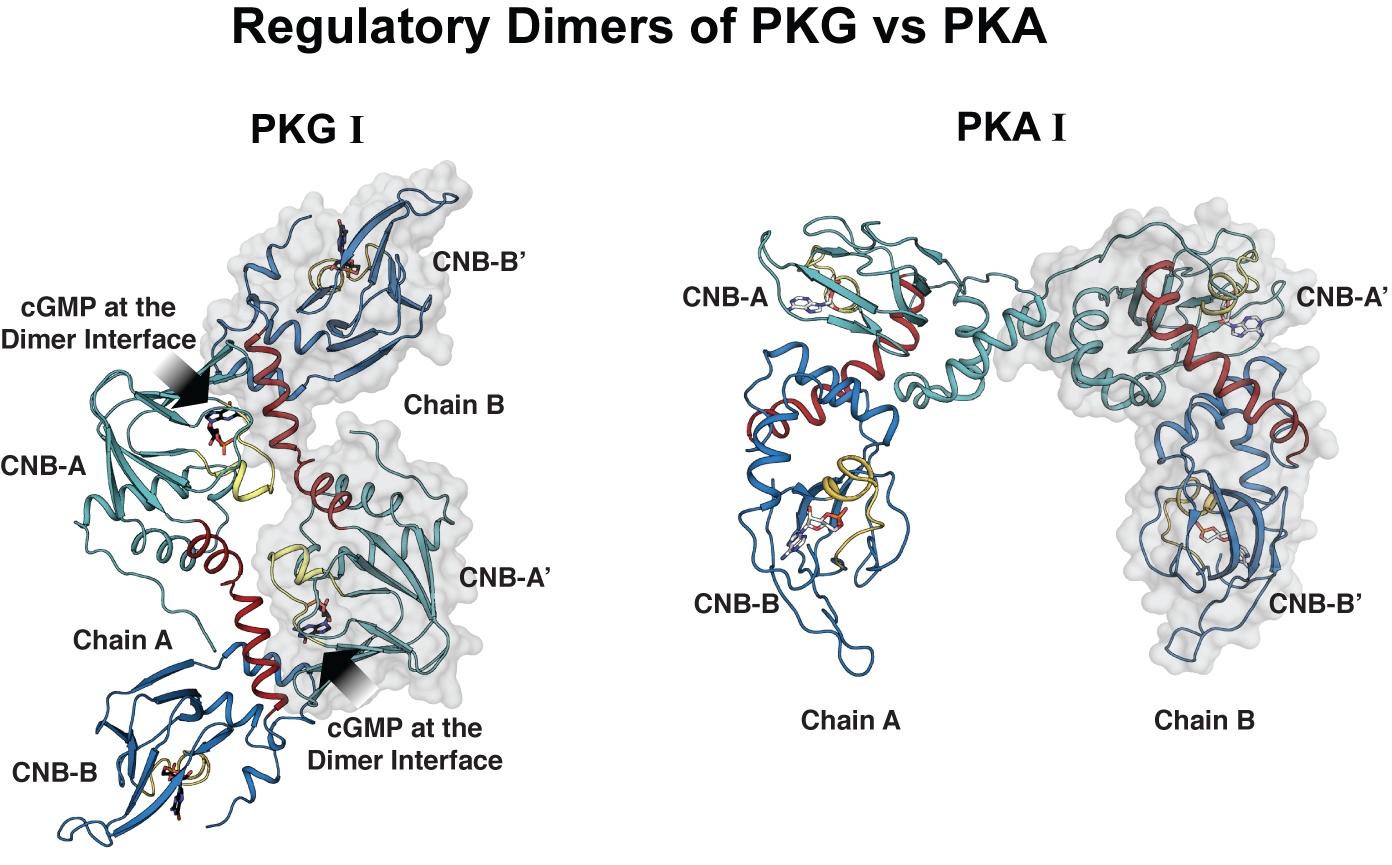
To study how PKGI regulates its activity, the researchers determined for the first time the crystal structure of the regulatory domain bound to cGMP, which is the activate form. They compared the crystal structure of PKGI with that of PKA.
In both PKA and PKG, the regulatory domains seem to form additional dimer contacts through their cyclic nucleotide binding domains, key domains that are critical for binding cyclic nucleotides for activation as well as for binding the catalytic domain for inhibition in the absence of cyclic nucleotides. But, in the case of PKG I, the crystal structure revealed that cGMP provides critical dimer contacts creating a new equilibrium species that may prevent the R- and C interaction.
“Having a regulatory domain that dimerizes through cGMP allows the regulatory domains to compete for each other and thus prevent this unnecessary R-C interaction. This can help sustain kinase activity critical for relaxing smooth muscle.”
Our study reveals a new activation mechanism for protein kinases, which we suggest to be considered in the design of drugs that target PKGI,” said Kim.
Other researchers who collaborated in this research include Jeong Joo Kim from the Department of Pharmacology at Baylor; Robin Lorenz from the Department of Biochemistry of the University of Kassel, Germany; Stefan T. Arold at the Computational Bioscience Research Center of King Abdullah University of Science and Technology in Saudi Arabia; Albert S. Reger from the Department of Pharmacology at Baylor and who is currently at Patheon Biologics-STL, St. Louis; Banumathi Sankaran from the Berkeley Center for Structural Biology, Lawrence Berkeley National Laboratory, Berkeley, California; Darren E. Casteel from the Department of Medicine, University of California, San Diego; and Friedrich W. Herberg from the Department of Biochemistry of the University of Kassel, Germany.
This project was supported by NIH grants R01 GM090161 and R21 HL111953, NCI (Y1-CO-1020), NIGMS (Y1-GM-1104), a grant from CCP4, and the STFC in the UK. In addition, this research received support from the King Abdullah University of Science and Technology (KAUST), the Federal Ministry of Education and Research Project NO PAIN (FKZ 0316177F) and the European Union (EU) FP7 collaborative project AFFINOMICS (contract no. 241481). The Berkeley Center for Structural Biology is supported in part by the NIH, the National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231. The SIBYLS beamline (ALS) is supported in part by US DOE program Integrated Diffraction Analysis Technologies (IDAT) and the NIH project MINOS (R01 GM105404).