Discovery of an underlying cause for infantile spasm points to a novel therapy
Infantile spasm (IS) is a severe epileptic syndrome of infancy that accounts for 50% of all epilepsy cases that occur in babies during the first year of life. Current treatment options for this disorder are limited and most affected infants grow up to have developmental delays, intellectual disabilities and other types of severe epilepsy.
A groundbreaking study, conducted in the laboratory of Dr. John Swann, professor of pediatrics – neurology at Baylor College of Medicine, director of the Gordon and Mary Cain Pediatric Neurology Research Foundation labs and investigator at the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, has the potential to transform the treatment landscape for babies with infantile spasms.

A few years back, Swann, a leading expert in epilepsy research, and his team pioneered discoveries that resulted in an FDA-approved treatment for severe epilepsy among tuberous sclerosis patients. He and his team have longstanding interest and experience in studying infantile spasms, an epileptic disorder diagnosed in roughly 2,500 babies in the United States each year.
Following the clues that point at the involvement of IGF-1 in infantile spasm
“It has been previously reported that infantile spasm patients with preexisting brain abnormalities have low levels of the hormone insulin-like growth factor-1 (IGF-1) in their cerebrospinal fluid. Based on that study, we were interested in investigating whether IGF-1 levels were altered in the brains of infantile spasm animal models and patients,” Swann said.
For their investigations, the team worked with a well-established animal model of infantile spasm that was developed in the Swann lab in 2008. The model mimics spasms that are virtually identical to those seen in patients with this disorder.
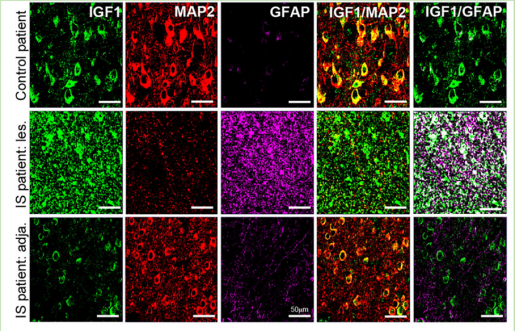
“As is expected after a brain injury, we saw an increase in IGF-1 levels in the non-neuronal support cells (glia) in the animal model. However, we were most intrigued by the remarkable and widespread decrease in IGF-1 expression in cortical neurons in brain regions adjacent to or further away from the affected glia — a phenomenon that we have never seen reported,” Swann said.
Next, the team studied resected cortical tissue from patients who had undergone surgery to control their intractable seizures. The results were remarkably similar to what they had seen in the animal model.
“More importantly, we found this reduction in cortical levels of IGF-1 had significant consequences in IS animal models because it dampened the overall activity of the IGF-1 molecular signaling pathways that regulate many important biological processes involved in early brain development and neuronal function,” said Dr. Carlos Ballester-Rosado, a postdoctoral associate in the Swann lab and first author of the study.

Treatment eliminates infantile spasms in animal model
To determine whether increasing IGF-1 levels in the cortex of IS animals could ameliorate spasms, the team used a smaller version of IGF-1, an IGF-1 analog that can cross the blood-brain barrier with greater ease than the full-length hormone. The IGF-1 analog they tested is a natural by-product of IGF-1 breakdown that is found normally in the brain. Moreover, this molecule has been previously demonstrated to successfully reverse behavioral defects in animal models of other neurodevelopmental disorders such as Rett syndrome and Phelan-McDermid syndrome.
“Using several lines of evidence, we first confirmed that this IGF-1 smaller molecule was capable of activating the IGF-1 signaling cascade in mice,” Swann said. “We then found — to our astonishment — that administration of the IGF-1 analog successfully eliminated spasms and an IS-specific chaotic brain activity pattern called hypsarrhythmia in most IS animals. We are excited because these findings raise the tantalizing possibility that this IGF-1 analog can be used to treat IS patients in the future.”
Read the report in full in the Annals of Neurology.
Others involved in the study are John Le, Trang Lam, Carrie Mohila, Sandi Lam, Anne Anderson and James Frost. The authors are affiliated with one or more of the following institutions: Baylor College of Medicine, Cain Foundation Laboratories and the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital.
The study was funded by CURE Epilepsy’s Infantile Spasms Initiative, grants from the National Institutes of Health (RO1 NS018309, RO1 NS105913 and R61/R33 NS112553) and Intellectual and Developmental Disabilities Centers (1U54 HD083092) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development.