Unraveling the complexity of vitamin B12 diseases
Vitamin B12, or cobalamin, is a dietary nutrient essential for normal human development and health and is found in animal-based foods but not in plant-based foods, unless they have been supplemented. Mutations in the genes encoding the proteins responsible for the metabolic processes involving vitamin B12 result in rare human inborn errors of cobalamin metabolism.


Vitamin B12 diseases can present a complex landscape of characteristics, and to better understand them Dr. Ross A. Poché, associate professor of molecular physiology and biophysics at Baylor College of Medicine, and his colleagues have studied two rare inherited vitamin B12 conditions that affect the same gene but are clinically distinct from the most common genetic vitamin B12 disorder.
Meet three distinct inherited vitamin B12 diseases
Patients with the most common inherited vitamin B12 disease, called cblC, suffer from a multisystem disease that can include intrauterine growth restriction, hydrocephalus (the build-up of fluid in the cavities deep within the brain), severe cognitive impairment, intractable epilepsy, retinal degeneration, anemia and congenital heart malformations. Previous work had shown that mutations in the MMACHC gene cause cblC disease.
It also was known that some patients presenting with a combination of typical and non-typical cblC characteristics do not have mutations in the MMACHC gene, but rather in genes that code for proteins called RONIN (also known as THAP11) and HCFC1. The resulting changes in these proteins lead to reduced MMACHC gene expression and a more complex cblC-like disease.
In this study, Poché and his colleagues looked for other genes that also might be affected by HCFC1 and RONIN gene mutations.
Tackling the complexity of two inherited vitamin B12 diseases
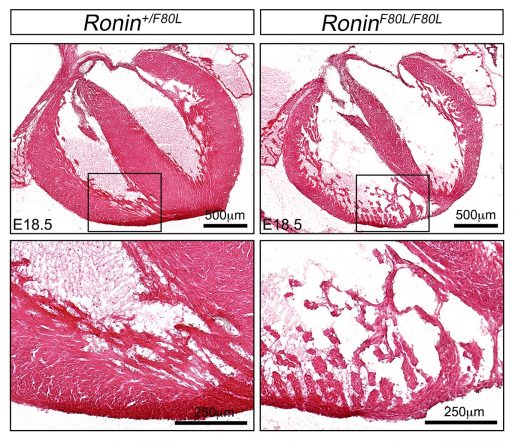
“We developed mouse models carrying the exact same mutations that the patients with cblC-like disease have in HCFC1 or RONIN genes, and recorded the animals’ characteristics,” Poché said. “We confirmed that they presented with the cobalamin syndrome as expected, but in addition we found that they had ribosome defects. Ribosomes form the protein-building machinery of the cell.”
This is the first time that the HCFC1 and RONIN genes have been identified as regulators of ribosome biogenesis during development.
The researchers demonstrate that this cblC-like disease affecting the function of RONIN and HCFC1 proteins is a hybrid syndrome as it is both a cobalamin disorder and a disease of ribosomes, or a ribosomopathy.
What the findings may mean for patients
The findings have potential therapeutic implications. “Some cblC-like patients may respond to some extent to cobalamin supplementation, but we anticipate that will not help the issues due to ribosome defects,” said Poché, member of the Dan L Duncan Comprehensive Cancer Center.
One step toward designing effective ribosomopathy therapies is to better understand what the defects in the ribosomes are. “We plan to functionally characterize the altered ribosomes at the molecular level to identify how their function is disrupted,” Poché said.

“There are many exciting aspects of this study, from the clinical implications to the basic science. The beauty is in how the work in patients is symbiotic with the work in the mouse model and how each system informs the other,” said co-author Dr. David S. Rosenblatt, professor in the departments of human genetics, medicine, pediatrics, and biology at McGill University and senior scientist at the Research Institute of the McGill University Health Centre.
Would you like to learn more about this study? Find it in the journal Nature Communications.
Other contributors to this work include co-first authors Tiffany Chern and Annita Achilleos, Xuefei Tong, Matthew C. Hill, Alexander B. Saltzman, Lucas C. Reineke, Arindam Chaudhury, Swapan K. Dasgupta, Yushi Redhead, David Watkins, Joel R. Neilson, Perumal Thiagarajan, Jeremy B. A. Green, Anna Malovannaya and James F. Martin. The authors are affiliated with one or more of the following institutions: Baylor College of Medicine; University of Nicosia Medical School, Cyprus; Michael E. DeBakey Veterans Affairs Medical Center, Houston; the Francis Crick Institute, London; King’s College London; McGill University Health Centre, Montreal and Texas Heart Institute, Houston.
This work was supported by the Dan L Duncan Comprehensive Cancer Center’s National Institutes of Health (NIH) award P30CA125123 for BCM Mass Spectrometry Proteomics Core, CPRIT Core Facility Award (RP170005) and the following NIH grants: R01 EY024906, R01 DE028298, T32 EY007102, T32 HL007676, R01 HL127717, R01 HL130804 and R01HL118761. Additional support was provided by the Vivian L. Smith Foundation, State of Texas funding and Foundation LeDucq Transatlantic Networks of Excellence in Cardiovascular Research (14CVD01).