A new mechanism of cisplatin resistance in cancer, and how to reverse it
Cancer cells repeatedly challenge researchers with their ability to develop resistance to treatments that were effective to begin with. Resistance to cisplatin, a platinum-based drug used as first-line chemotherapy treatment is a prominent example that has been extensively studied in the lab of Dr. Weei-Chin Lin, professor of medicine – hematology and oncology and of molecular and cellular biology at Baylor College of Medicine.
“Cisplatin is a common chemotherapeutic drug and is widely used to treat solid tumors, such as those in the ovaries, testes, cervix, bladder, lungs, breast, head and neck and others,” said Lin, a member of Baylor’s Dan L Duncan Comprehensive Cancer Center. “Despite its effectiveness, many patients eventually develop resistance to cisplatin, which presents a major obstacle to their treatment.”

Cisplatin delivers its attack by entering cancer cells and binding to and damaging DNA, which can lead to cell death. Cells respond to DNA damage by triggering DNA repair mechanisms that involve a number of proteins. Previous studies support that enhanced DNA repair seems to be the most likely cause of cisplatin resistance.
Enter ACTL6A, a gene involved in remodeling chromatin, the material that forms the chromosomes and consists of protein, RNA and DNA. In addition, the ACTL6A gene is frequently amplified in cancer. According to the Cancer Genome Atlas, this gene is overexpressed in 37.4% of lung squamous cell carcinoma, 19.5% of ovarian cancer and 17.6% of esophageal cancer.
Lin and his colleagues hypothesized that, given its overexpression in many cancer types and its connection to chromatin remodeling, the ACTL6A protein might be a good candidate as a mediator of cisplatin resistance.
“We wanted to know whether amplification of gene ACTL6A would result in cisplatin resistance,” Lin said. “If it were involved, then we could study the mechanism and explore ways to counter it.”
Connecting ACTL6A with cisplatin resistance
Dr. Yang Xiao, a postdoctoral associate in medicine – hematology and oncology working in the Lin lab, took on the challenge of investigating the possible connection of ACTL6A protein with cisplatin resistance.
Xiao began her study by looking into several cancer genome databases to assess whether having high expression of ACTL6A was linked to patient outcome and response to chemotherapy.
“Indeed, we found that high expression of ACTL6A not only was associated with shorter patient lifespan in both lung and ovarian cancer, but also that cancers resistant to cisplatin treatment expressed higher levels of ACTL6A than cancers sensitive to cisplatin,” Xiao said. “We also observed this association between higher levels and resistance to cisplatin in cell lines of different types of cancers grown in the lab.”
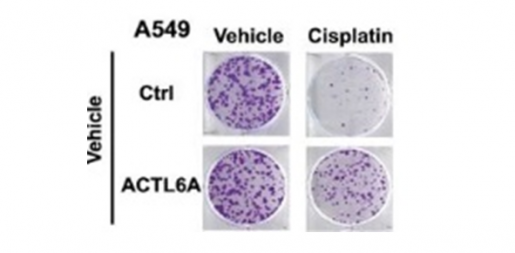
To further explore this connection, Xiao and Lin then tested the effect of experimentally overexpressing the gene on cisplatin resistance in a panel of lung cancer cells lines. “We found that overexpression did not affect cancer cell growth, but significantly increased cell survival after cisplatin treatment. Supporting this fact, we found that removing the ACTL6A gene from the cells made them more susceptible to cisplatin treatment,” Lin said. “We observed similar results in experiments conducted in animal models.”
How ACLT6A makes cancer cells resistant to cisplatin and how to reverse it
Knowing that the ACTL6A protein binds to histones, proteins associated with DNA, and seemed to be involved in chromatin repair, Xiao and Lin tested whether it promoted cisplatin resistance by affecting cisplatin-mediated DNA damage and/or by enhancing the repair of cisplatin-mediated damage.
ACTL6A overexpression did not prevent cisplatin-induced DNA damage, but it stimulated its repair,” Xiao said.
“Other experiments showed that ACTL6A-enhanced DNA repair involved epigenetic or chemical modifications of the chromatin remodeling complex SWI/SNF.”
“These findings gave us the idea that blocking or disturbing the epigenetic changes in the chromatin complex could help counter the resistance to cisplatin,” Lin said.
“We tested a histone deacetylase (HDAC) inhibitor, panobinostat, which could interfere with the epigenetic changes caused by ACTL6A overexpression. Panobinostat is an FDA-approved drug currently used only for multiple myeloma, but not in other clinical settings yet,” Lin said.
The results were encouraging. In the presence of panobinostat, ACTL6A could no longer promote cisplatin resistance. Panobinostat resensitized ACTL6A-overexpressing cells to cisplatin, both in cells grown in the lab and in animal models.
This work shows a new mechanism of cisplatin resistance in cancer caused by a commonly amplified gene,” Lin said. “Importantly, we identified panobinostat, an HDAC inhibitor, as a potential therapeutic reagent that can reverse cisplatin resistance caused by ACTL6A overexpression.”
“Further studies are required before our findings can be translated into clinical applications, but we hope that our work will provide a stimulus and scientific rationale for oncologists to incorporate HDAC inhibitors into future clinical trials to overcome platinum resistance in many types of cancer.”
Are you interested in all the details of this work? Find it in the Proceedings of the National Academy of Sciences, U.S.A.
Fang-Tsyr Lin at Baylor College of Medicine, also contributed to this work.
This work was supported by NIH grant R01CA100857, Rivkin Center for Ovarian Cancer Pilot Award and R01CA203824, Department of Defense Grants W81XWH-18-1-0329 and W81XWH-19-1-0369 and T32 Fellowship T32CA174647.