
A new gut-brain connection that keeps weight off (or on) the scale
Eating extra servings typically shows up on the scale later, but how this happens has not been clear. In his lab at Baylor College of Medicine, Dr. Makoto Fukuda and his colleagues investigate how the body manages energy balance and affects weight. They have discovered a new gut-brain connection that provides a better understanding of how those extra servings lead to weight gain.


Leptin, a hormone produced by fat cells, is important in the control of body weight both in humans and mice. Leptin works by triggering in the brain the sensation of feeling full when we have eaten enough, and we stop eating. However, in obesity resulting from consuming a high-fat diet or overeating, the body stops responding to leptin signals – it does not feel full, and eating continues, leading to weight gain.
“We didn’t know how a high-fat diet or overeating leads to leptin resistance,” said Fukuda, assistant professor of pediatrics at Baylor and the USDA/ARS Children’s Nutrition Research Center at Baylor and Texas Children’s Hospital. “My colleagues and I started looking for what causes leptin resistance in the brain when we eat fatty foods. Using cultured brain slices in petri dishes we screened blood circulating factors for their ability to stop leptin actions. After several years of efforts, we discovered a connection between the gut hormone GIP and leptin.”
Uncovering the GIP-leptin connection
GIP is one of the incretin hormones produced in the gut in response to eating and known for their ability to influence the body’s energy management. To determine whether GIP was involved in leptin resistance, Fukuda and his colleagues first confirmed that the GIP receptor, the molecule on cells that binds to GIP and mediates its effects, is expressed in the brain.
Then the researchers evaluated the effect blocking the GIP receptor would have on obesity by infusing directly into the brain a monoclonal antibody developed by Dr. Peter Ravn at AstraZeneca that effectively prevents the GIP-GIP receptor interaction. This significantly reduced the body weight of high-fat-diet-fed obese mice.
“The animals ate less and also reduced their fat mass and blood glucose levels,” Fukuda said. “In contrast, normal chow-fed lean mice treated with the monoclonal antibody that blocks GIP-GIP receptor interaction neither reduced their food intake nor lost body weight or fat mass, indicating that the effects are specific to diet-induced obesity.”
Further experiments showed that if the animals were genetically engineered to be leptin deficient, then the treatment with the specific monoclonal antibody did not reduce appetite and weight in obese mice, indicating that GIP in the brain acts through leptin signaling. In addition, the researchers identified intracellular mechanisms involved in GIP-mediated modulation of leptin activity.
“In summary, when eating a balanced diet, GIP levels do not increase and leptin works as expected, triggering in the brain the feeling of being full when the animal has eaten enough and the mice stop eating,” Fukuda said. “But, when the animals eat a high-fat diet and become obese, the levels of blood GIP increase. GIP flows into the hypothalamus where it inhibits leptin’s action. Consequently, the animals do not feel full, overeat and gain weight. Blocking the interaction of GIP with the hypothalamus of obese mice restores leptin’s ability to inhibit appetite and reduces body weight.”
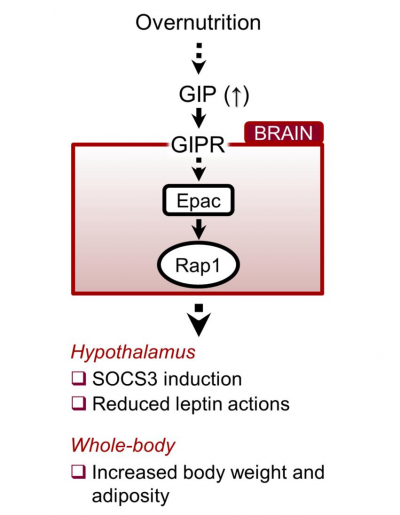
These data indicate that GIP and its receptor in the hypothalamus, a brain area that regulates appetite, are necessary and sufficient to elicit leptin resistance.
This is a previously unrecognized role of GIP on obesity that plays directly into the brain.
Although more research is needed, the researchers speculate that these findings might one day be translated into weight loss strategies that restore the brain’s ability to respond to leptin by inhibiting the anti-leptin effect of GIP.
Find the complete study in the Journal of Clinical Investigation.
Other contributors to this work include Kentaro Kaneko, Yukiko Fu, Hsiao-Yun, Lin, Elizabeth L. Cordonier, Qianxing Mo, Yong Gao, Ting Yao, Jacqueline Naylor, Victor Howard, Kenji Saito, Pingwen Xu, Siyu S. Chen, Miao-Hsueh Chen, Yong Xu, Kevin W. Williams and Peter Ravn. For author affiliations refer to the published paper.
This work was supported by USDA CRIS 6250-51000-055, AHA-14BGIA20460080, NIH-P30-DK079638, NIH R01DK104901, AHA-15POST22500012, the Uehara Memorial Foundation 201340214, NIH-T32HD071839, AHA-13POST13800000 and AHA-15POST22670017. Further support was provided by NIH R01DK100699, DK119169, China Scholarship Council 201406280111, CRIS 6250-51000-059 and NIH P30-DK079638. This project was also supported in part by the Genomic and RNA Profiling Core at Baylor College of Medicine with funding from P30 Digestive Disease Center Support Grant (NIDDK-DK56338) and P30 Cancer Center Support Grant (NCI-CA125123).


