Bacteria can have a ‘sweet tooth,’ too
Clostridium difficile infections have always been a problem in hospitals, but during the last 15 years they have become the most common cause of hospital-acquired infections in developed countries. This problem is one of the research interests of the lab of Dr. Robert Britton where researchers have discovered what seems to have prompted C. difficile to become a superbug.
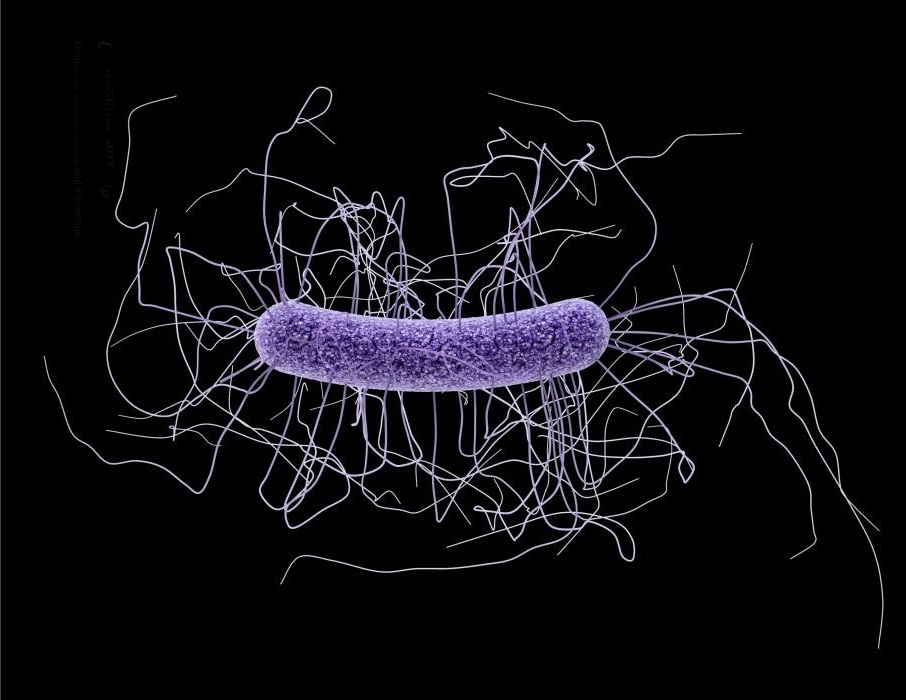
“Our group and others have found that C. difficile lineages RT027 and RT078 have become dominant more recently around the globe,” said Dr. James Collins, a postdoctoral associate in the Britton lab. “These lineages have been present in people for years without causing major outbreaks. In the 1980s they were not epidemic or hypervirulent, but after the year 2000 they began to predominate and cause major outbreaks. We wanted to know what had helped these lineages become a major health risk.”

According to the Center for Disease Control and Prevention, C. difficile is a major cause of infectious disease-related death in the United States. In 2015, the CDC reported that C. difficile caused almost a half-million infections in patients in a year and 29,000 estimated deaths. The bacteria cause life-threatening inflammation of the colon and diarrhea. Patients 65 years and older are at most risk, and most infections occur in people who have received medical care and antibiotics.
Diet and bacterial virulence
Resistance to fluoroquinolone antibiotics is likely one of the factors that is helping lineage RT027 cause epidemics.
“However, fluoroquinolone resistance is also a characteristic of other C. difficile lineages that are not epidemic,” Collins said. “We searched for other factors that would promote the virulence of RT027 and RT078.”
The researchers investigated what sources of food RT027 and RT078 preferred. They discovered that these lineages can grow on levels of sugar trehalose that are about 1,000 times lower than those needed by other lineages of these bacteria, giving RT027 and RT078 a major advantage. Each lineage is highly efficient at using trehalose and evolved independent mechanisms to utilize this sugar. To connect the ability to metabolize low levels of trehalose with increased disease severity, the researchers worked with a mouse model of C. difficile infection.
“Mice received a strain of the RT027 lineage of C. difficile and a diet with or without low trehalose levels,” Collins said. “What the mice ate made a difference to the virulence of the infection; mortality was higher in the group consuming trehalose.”
Further experiments showed that increased disease severity in the presence of trehalose could not be explained by the mice having higher numbers of bacteria. Instead, what made the disease more severe was that RT027 produced higher levels of toxins.
These and other experiments provide evidence that dietary trehalose has contributed to the predominance of epidemic C. difficile lineages and to their virulence. Because the genetic factors that allow these bacteria to metabolize trehalose and increase the production of toxins were present well before the outbreaks started, the researchers investigated what could have triggered the epidemics.

“In 2000, trehalose was approved as a food additive in the United States for a number of foods from sushi and vegetables to ice cream. About three years later, the reports of outbreaks with these lineages started to increase,” said Britton, professor of molecular virology and microbiology and member of the Alkek Center for Metagenomics and Microbiome Research and the Dan L Duncan Comprehensive Cancer Center at Baylor College of Medicine. “Other factors may also contribute, but we think that trehalose is a key trigger.”
“An important contribution of this study is the realization that what we once considered a perfectly safe sugar for human consumption, can have unexpected consequences,” Collins said. “Our study suggests that the effect of trehalose in the diet of patients in hospitals with RT027 and RT078 outbreaks should be further investigated.”
Find all the details of this study in the journal Nature.
Other contributors to this work include H. Danhof and J.M. Auchtung at Baylor College of Medicine; C. Robinson at the University of Oregon; C.W. Knetsch and H.C. van Leeuwen at Leiden University Medical Center in The Netherlands and T.D. Lawley at Wellcome Trust Sanger Institute in the United Kingdom.
Financial support was provided by the National Institutes of Health grants U01AI124290-01 and 5U19AI09087202.
-By Ana María Rodríguez, Ph.D.