Small brain region linked to nicotine addiction plays a major role in the control of appetite
[See video beolow]
Researchers studying the mystery of how the brain controls appetite have brought into play a small region of the brain, the basal forebrain. In this region, a small group of a few thousand acetylcholine-producing cells moderates whether an individual is hungry or full. In mouse studies, when these cells are impaired, the animals eat constantly and become obese. But, when the cells enhance their activity, the mice eat little and lose weight.
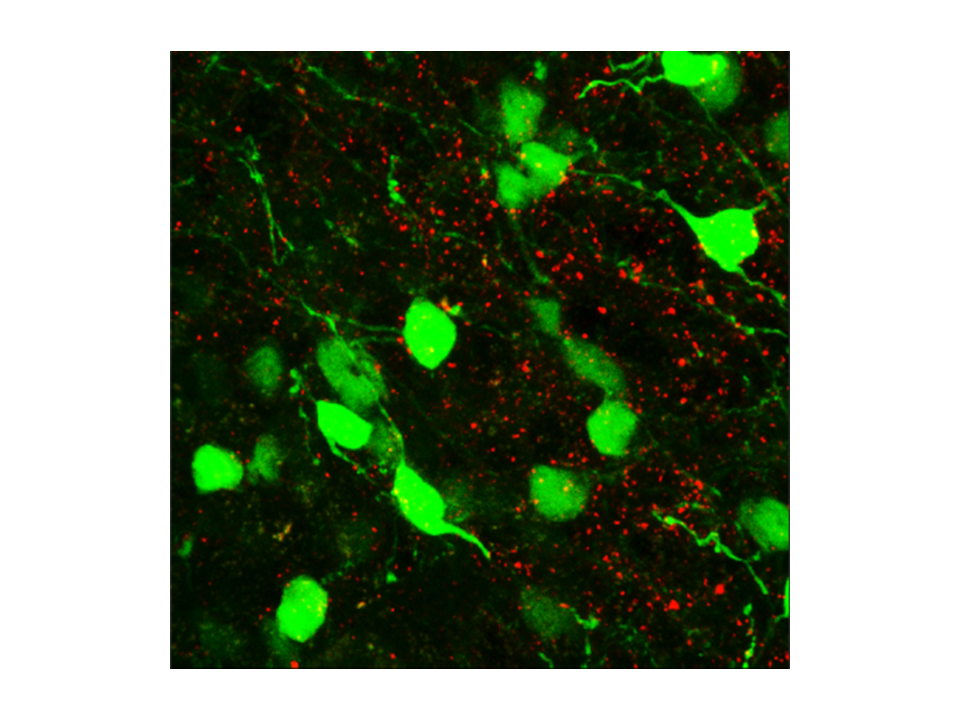
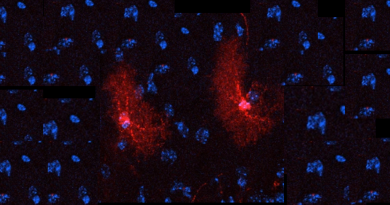
The team of researchers, who published the study in Nature, think that this finding opens the possibility of developing drugs to help treat obesity, a major health problem around the world, as well as other eating disorders.
When serendipity knocks
“When we started these experiments, we were not looking for feeding-related circuits at all,” said senior author Dr. Benjamin Arenkiel, an associate professor of molecular and human genetics and of neuroscience at Baylor College of Medicine.

Arenkiel and colleagues were studying how neurons form connections with each other – how they build synapsis – in the basal forebrain. They knew from experiments with neurons in laboratory cultures that adding acetylcholine triggers synapsis growth in these cells. To study the effect of acetylcholine in a living mouse, the scientists made genetic modifications that eliminated only the cells that produce acetylcholine in the basal forebrain.
“After several weeks, we observed a totally unexpected result. The mice lacking acetylcholine in the basal forebrain were almost four times the size of a normal mouse,” said Arenkiel, who is also a McNair Scholar at Baylor and an investigator at the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital. “It turned out that acetylcholine produced by neurons in the basal forebrain is regulating an obesity circuit.”
Arenkiel and colleagues then decided to follow this new clue and refocused their research to study how acetylcholine regulates appetite.
“Without acetylcholine, the mice seem hungry all the time; they are insatiable,” said Arenkiel. “If you limit the amount of food, they will lose weight equally to the mice with acetylcholine in their basal forebrain.”
When the researchers increased the production of acetylcholine in the basal forebrain in normal mice, the animals seemed to have lost their appetite. “They won’t starve themselves; they will be almost anorexic. We think acetylcholine-producing cells in the basal forebrain are regulating the satiety cues in the brain.”
The nicotine link
“Nicotine, known for attenuating the appetite, works like acetylcholine does. Both molecules bind to the same molecules or receptors on the surface of cells,” said Arenkiel. “The brain circuit that controls feeding also interfaces with nicotine and nicotine addiction. This suggests that it might be possible in the future to use currently available nicotine receptor agonists to treat obesity.” Nicotine receptor agonists are drugs that bind to nicotine receptors and mimic the action of acetylcholine.
“This study has allowed us to better understand the brain circuit that controls appetite,” said Arenkiel. “The hypothalamus receives clues from the senses; for instance, the smells and appetizing looks of homemade food. It also receives internal clues, such as the levels of blood sugar and insulin in the body. The hypothalamus processes all these information to regulate feeding.”
“What we have found here is that acetylcholine can influence the hypothalamic circuit and regulate satiety,” said Arenkiel.
###
Other authors that contributed to this work include Alexander M. Herman, Joshua Ortiz-Guzmán, Mikhail Kochukov, Isabella Herman, Katie Quast, Jay M. Patel, Burak Tepe, Jeffrey C. Carlson, Kevin Ung, Jennifer Selever and Qingchun Tong. The researchers are affiliated with one or more of the following institutions, Baylor College of Medicine, Texas Children’s Hospital and the University of Texas Health Science Center in Houston.
This study received financial support from NIH grants 5F31NS089411, R01NS078294, and R01DK109934, the Klarman Family Foundation, the Klingenstein-Simons Fellowship Award, the Brain and Behavior Research Foundation, the Charif Souki Fund, and the McNair Medical Institute. The authors further acknowledge the BCM Diabetes and Endocrinology Research Center and the services of its Mouse Metabolism Core supported by NIH grant P30DK079638, and the BCM-IDDRC U54HD083092.