Mapping functional relations among proteins and genes helps illuminate their role in cancer
Large-scale protein and gene profiling have massively expanded the landscape of cancer-associated proteins and gene mutations, but it has been difficult to discern whether they play an active role in the disease or are innocent bystanders. In a study published in Nature Cancer, researchers at Baylor College of Medicine revealed a powerful and unbiased machine learning-based approach called FunMap for assessing the role of cancer-associated mutations and understudied proteins, with broad implications for advancing cancer biology and informing therapeutic strategies.

“Gaining functional information on the genes and proteins associated with cancer is an important step toward better understanding the disease and identifying potential therapeutic targets,” said corresponding author Dr. Bing Zhang, professor of molecular and human genetics and part of the Lester and Sue Smith Breast Center at Baylor.
Mapping functional relations among proteins helps illuminate their role in cancer
“Our approach to gain functional insights into these genes and proteins involved using machine learning to develop a network mapping their functional relationships,” said Zhang, member of Baylor’s Dan L Duncan Comprehensive Cancer Center and a McNair Scholar.
It’s like, I may not know anything about you, but if I know your LinkedIn connections, I can infer what you do.”
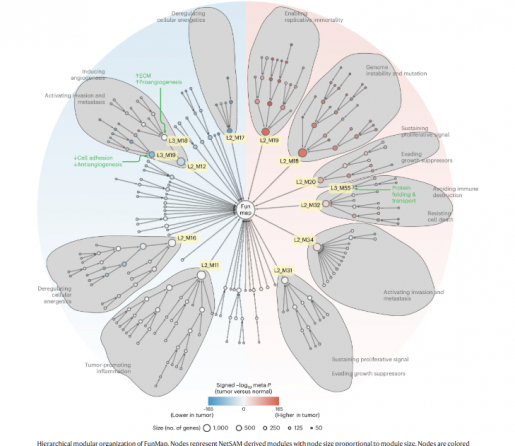
The team developed FunMap, a functional network of 10,525 genes constructed using a supervised machine learning method that integrates protein datasets and RNA sequencing data from 11 cancer types recently harmonized by the Clinical Proteomic Tumor Analysis Consortium (CPTAC) pan-cancer working group.
“With FunMap, we found 196,800 associations among 10,525 proteins – a comprehensive and unbiased proteomic coverage and a high level of functional relevance,” Zhang said. “Two key differences between our approach and previous gene co-expression network studies are first, the integration of cancer protein data with mRNA expression data and second, the application of supervised machine learning to synergize all datasets to maximize the predictive power.
Unexpectedly, our approach outperformed protein–protein interaction networks in discriminating between functionally relevant and irrelevant gene pairs.”
Through network analysis, FunMap uncovers protein modules and a hierarchical modular organization linked to cancer hallmarks and clinical characteristics, predicts the functions of understudied cancer proteins, offers deeper insights into established cancer drivers and identifies drivers with low mutation frequency.
More than 200 genes are highly overexpressed or under-expressed in cancer, but we know very little about their specific roles in the disease,” Zhang said. “When we mapped these genes in our network, we were able to look at the neighborhood and make a prediction about their function.”
For example, the expression of the understudied gene MAB21L4 is significantly below normal in three types of cancer tumors. FunMap showed that this gene’s network neighborhood is enriched for genes associated with epithelial cell differentiation, the suppression of which plays a critical role in tumor progression. Clinical tumor grading data, together with a recent study showing that loss of MAB21L4 blocks differentiation to drive the development of squamous cell carcinoma, provide strong evidence to support a tumor suppressor role of MAB21L4.
FunMap can reveal unrecognized cancer drivers
Moreover, leveraging cutting edge deep learning methods with FunMap uncovered numerous previously unrecognized cancer drivers with low mutation frequencies, including a novel tumor suppressor role for LGI3, supported by gene knockout experimental data.
This study highlights the great potential of integrating machine learning and proteogenomic profiling to gain a deeper understanding of complex cancer systems. By generating a comprehensive functional network, this approach provides a robust framework for cancer functional genomics research, offering valuable insights into mutations and cancer-associated proteins.
“These findings can greatly aid in prioritizing targets for clinical translation, ultimately contributing to the development of more effective cancer therapies,” Zhang said.
The FunMap Python package is a fully open source available for download from the Python Package Index (https://pypi.org/project/funmap).
Co-first authors, Zhiao Shi and Jonathan T. Lei, and John M. Elizarraras, also contributed to this work. All are affiliated with Baylor College of Medicine.
The authors acknowledge contributions from the CPTAC and its Pan-Cancer Analysis working group. This work was supported by National Institutes of Health grants from the National Cancer Institute (U24 CA210954, U24 CA271076, R01 CA245903 and U01 CA271247), the Cancer Prevention and Research Institute of Texas (CPRIT) (award RR160027), a CPRIT scholarship and a cancer scholarship by the McNair Medical Institute at the Robert and Janice McNair Foundation.
Follow From the Labs on X @BCMFromtheLabs and Instagram!