Using the power of social media to solve a genetic mystery

By Ruth SoRelle, M.P.H.
“Help us find others like Tess.” Bo Bigelow’s plea jumps off the page of his blog, echoing across the continent from his leafy green home city of Portland, Maine.
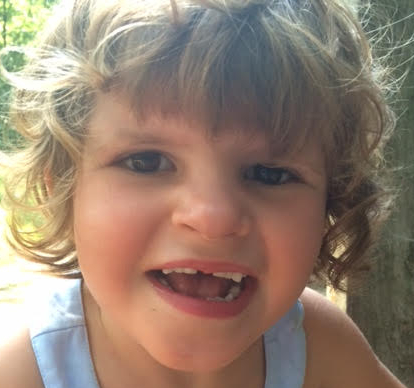
Tess is his curly-headed 5-year-old daughter, now missing her first tooth. Bigelow, left the law to be a stay-at-home dad to special-needs Tess and her older brother Dana.
Tess has a mutation in a gene called USP7. Finding more patients with a mutation in the same gene will help unravel the mystery of her disease. Her parents are relying on the power of the internet to help solve her problem.
Call to action
When he posted his call to action, all Bigelow knew was that his young daughter has a mutation in her USP7 gene and that she has global developmental delay, hip dysplasia and visual impairment caused by her brain, not a problem in her eyes themselves – among other problems. An article in the New Yorker magazine by Seth Mnookin gave him hope that finding other children with the same mutated gene as his daughter might point the way toward understanding what was causing her problems and even spark the science that could solve.
Unbeknownst to Bigelow, in a laboratory in steamy Houston and in collaboration with a researcher at The University of Texas Southwestern School of Medicine, a young researcher named Dr. Christian Schaaf was already on the trail of USP7, and he had found seven children with USP7 mutations and symptoms similar to those of Tess. Schaaf sought out Dr. Ryan Potts of the UT Southwestern School of Medicine at Dallas, who had already been studying the gene.
Becoming a disease gene
In a report in the journal Molecular Cell, Schaaf, Potts, and their colleagues (including first authors Yi-Heng Hao of UT Southwestern and Michael D. Fountain Jr., of Baylor College of Medicine) describe how USP7 becomes a “disease gene.” It begins with the triad of genes – MAGEL2, TRIM27 and USP7 – and their effect on the critical process of the recycling of proteins within cells that is regulated by ubiquitination and deubiquitination.
“This process keeps everything in homeostasis,” said Schaaf, an assistant professor of molecular and human genetics at Baylor and the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital. “It makes sure the right proteins are recycled at the right pace. We began to look at USP7 because we realized it interacts closely with MAGEL2. If you lose the protein MAGEL2, it causes Schaaf-Yang syndrome,” a disorder discovered by Schaaf and his colleague Dr. Yaping Yang that has symptoms similar to those seen in the disorder suffered by Tess and the seven youngsters that Schaaf had located through a search of genome databases.

Protein complex
Proteins associated with USP7, MAGEL2 and TRIM27 form a complex that regulates each other and those downstream of their activity within the cell.
Basically, players in the complex act like a rheostat, fine-tuning the protein recycling pathway.
“It’s like the thermostat on an air conditioner that self regulates to keep the temperature constant,” said Schaaf. “This process keeps everything in balance. If you lose the function of MAGEL2, the disease called Schaaf-Yang results. If you lose the function of USP7, it may cause a disease that looks very similar – molecularly and clinically.”
After Schaaf had sent his paper in to the journal, students in his lab became aware of Bigelow and his blog that spearheads the search for help for his curly-headed daughter whose USP7 gene is awry.

Contact between researcher and parents
They have contacted Bigelow and Tess’ data may become part of future studies.
It’s a new method of finding patients with mutated genes that is becoming increasingly important in the study of genetics. Driven by technology that makes it possible to determine the sequence of an individual’s exome and look for mutations, often by comparing it to the exomes of the two parents, parents and patients often take to social media in order to find answers to their problems. Bigelow hoped his blog and podcasts about Tess, “Stronger Every Day,” would draw parents with similar dilemmas to him. As their numbers grow, they can push for research into the problem that affects their children.
“I’ve taken the strategy of the guy in the New Yorker article,” he said. “Let other people find out. It’s a reverse beacon that so far has connected us with this study and Dr. Schaaf. I would love to find more people.”
“You hear the term ‘rare disease,’ and I’m not sure it’s the best name for it,” said Bigelow. “If you look at all the so-called rare disease, it appears that there may be as many 39 million people afflicted with them. The idea that you are along is fading away as we connect with people.”
Others who took part in this work include Fan Xia, Weimin Bi, Sung-Hae L. Kang, Ankita Patel and Jill A. Rosenfeld, all of Baylor; Klementina Fon Tacer of UTSW in Dallas; Cedric Le Caignec and, Bertrand Isidor of Service de Genetique Medicale at CHU de Nantes, Nantes, France; Ian D. Krantz and Sarah E. Noon of Children’s Hospital of Philadelphia; Jean P. Pfotenhauer and Thomas M. Morgan of Vanderbilt University School of Medicine in Nashville, Tenn.; Rocio Moran of Cleveland Clinic in Ohio; Robert C. Pedersen of Tripler Army Medical Center in Honolulu, Hawaii; and Margarita S. Saenz of Children’s Hospital Colorado in Aurora.
Funding for this work came from Michael L. Rosenberg Scholar in Medical Research fund; the Cancer Prevention and Research Institute of Texas (Grant R1117), the Welch Foundation (Grant I-1821), National Institutes of Health (Grant R01GM111332), the Joan and Stanford Alexander Family and the Foundation for Prader-Willi Research. The project was supported in part by Intellectual and Developmental Disabilities Research Center at Baylor (Grant 1U54 HD083092 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development).