Missing “copy” of Jag1 gene results in mouse model for Alagille syndrome
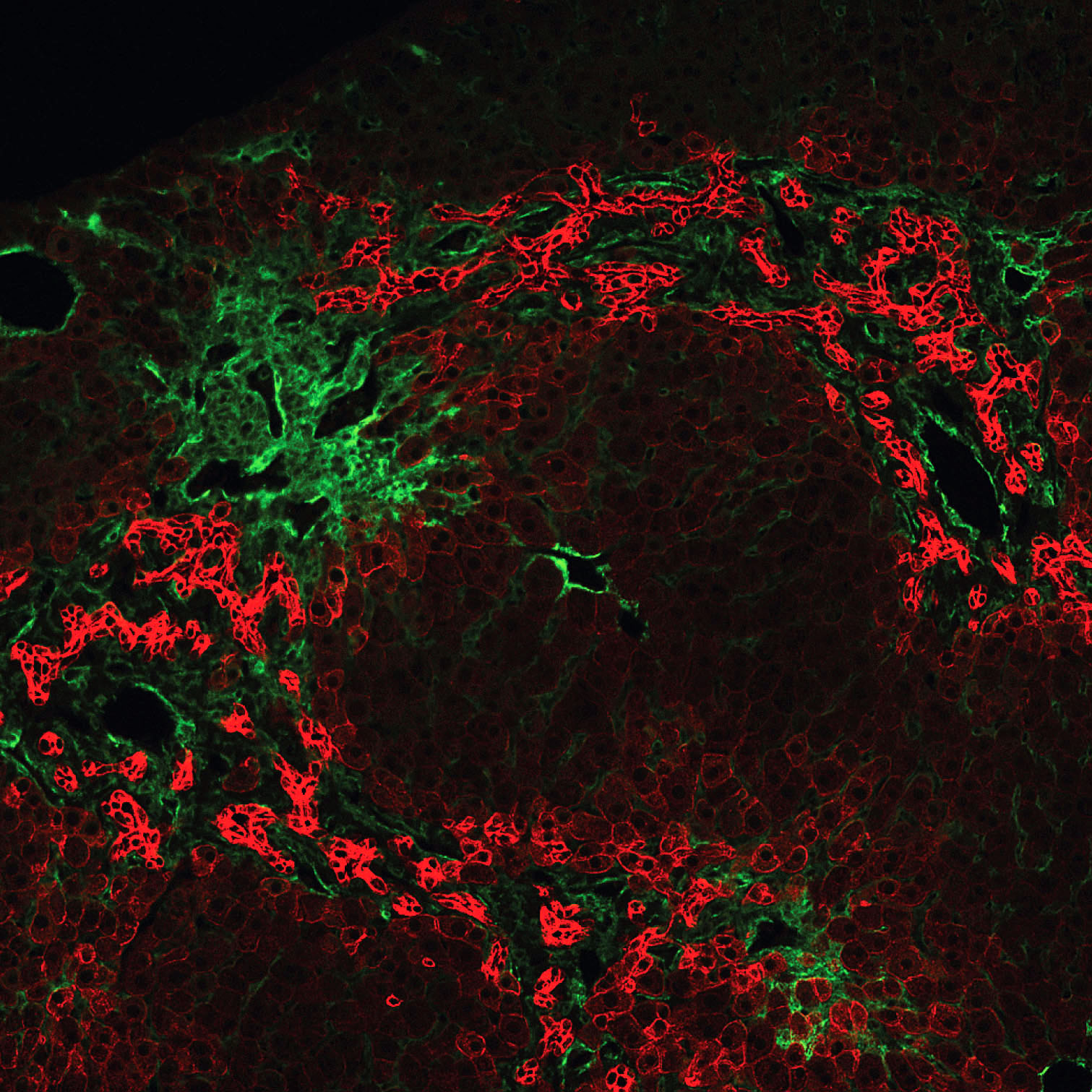
extensive ductular reactions in the periportal areas.
By Julia Parsons
When mice usually used in studies of genetic disease lack a copy of the jagged gene (Jag1), they have many of the characteristics, including bile duct abnormalities, of human Alagille syndrome, a developmental disorder characterized by bile duct and cardiovascular defects, as well as abnormalities in other organs, reported Baylor College of Medicine experts in a report that appears in the journal Hepatology.
Research conducted 20 years ago on Alagille syndrome patients showed that the disease is caused by loss of one copy of JAG1, but subsequent studies on Jag1 heterozygous mice did not report any outward characteristics of phenotype of Alagille syndrome other than eye abnormalities.
A different genetic background is one in which other related genes interact and affect the function of the gene being studied.
Bile duct defects

When Jag1 heterozygous mice are maintained on a different genetic background than was used in that original study, they show bile duct defects similar to Alagille syndrome,” said Dr. Shakeel M. Thakurdas, postdoctoral fellow in the department of molecular and human genetics at Baylor and lead author on the paper. “Our study suggests that on a C57BL/6 genetic background (which refers to a type of mouse commonly used in the laboratory) Jag1 heterozygous mice (which mean they have one normal or wild-type Jag1 gene and one that is mutated) can serve as a model for the bile duct abnormalities of Alagille syndrome.”
“Our work started with the assumption, based on previous studies, that Jag1 heterozygous mice do not have bile duct phenotypes; however, we stumbled on bile duct defects in these mice,” said Dr. Hamed Jafar-Nejad, associate professor in the department of molecular and human genetics and the Program in Developmental Biology at Baylor and corresponding author.

Improving survival, phenotypes
“Our work shows that simultaneous removal of one copy of a second gene, Poglut1, improves the survival and bile duct phenotypes in this model,” Thakurdas said.
The study shows that the JAG1 protein is a target of POGLUT1, an enzyme that adds carbohydrate residues to a number of proteins in humans and other animals.
“We hope that the establishment of a representative mouse model for Alagille syndrome and identification of a glycosyltransferase enzyme as a dominant genetic suppressor of the phenotypes will help us better understand the pathophysiology of bile duct abnormalities in Alagille syndrome,” Jafar-Nejad said. “It might also offer a potential path for therapeutic intervention.”
Studies receive award
In recognition of the potential importance of these studies for Alagille syndrome research, Jafar-Nejad recently received an Alagille Syndrome Accelerator Award from The Medical Foundation, a division of Health Resources in Action.
Others who took part in this research include Mario F. Lopez and Neda Zarrin-Khameh from Baylor College of Medicine; Rodrigo Fernandez-Valdivia from Wayne State University, Detroit, MI (a former postdoc in Jafar-Nejad lab); and Shinako Kakuda and Robert S. Haltiwanger from Stony Brook University, Stony Brook, NY (currently at the Complex Carbohydrate Research Center, University of Georgia).
Funding for this work came from Cell Lineage and Differentiation Research Grant No. 1-FY10-501 and the Basil O’Connor Starter Award No. 5-FY07-654 from the March of Dimes Foundation (H.J.-N.), and R01GM084135 (H.J.-N.) and R01GM061126 (R.S.H.) from the National Institutes of Health.